Introduction to the host-microbial interactions in periodontal diseases
The microbial biofilm that forms around the teeth is the main cause of periodontal disease initiation and progression. This biofilm is a complex community of microorganisms which produces various virulence factors that initiate the inflammatory response. The enzymes released by bacteria in the biofilm include proteases that are capable of disintegrating collagen, elastin, fibronectin, fibrin and various other components of the intercellular matrix of both epithelial and connective tissue. Other proteases are leukotoxins which are capable of killing leukocytes. Endotoxins are produced by Gram-negative bacteria which are strong inducers of cytokine production. The term lipopolysaccharide (LPS) is often used interchangeably with endotoxin. As the subgingival biofilm is majorly composed of Gram-negative bacteria, endotoxins produced by them leads to the induction of cytokine production by host cells, which causes inflammatory changes, like increased vascular permeability and engorgement of blood vessels. In the present discussion, we shall read about various interactions that take place between the plaque microbiota and the host immune response.
Evolution of our understanding of periodontal disease progression
Our understanding about periodontal disease progression improved tremendously during 1960’s, when animal and human experiments demonstrated the role of bacteria in the initiation of gingivitis and periodontitis 1, 2. These studies led to the proposal of the model of bacterial etiology of periodontal diseases. Further investigations in this field led to the advancement in our knowledge of pathogenic bacteria causing disease progression. Specific Gram-negative, anaerobic, or microaerophilic bacteria were implicated in the causation of periodontitis 3-7. During the late 1970’s and early 1980’s protective and destructive roles of the immunoinflammatory responses were described in health and disease 8-14.
Most of the models of periodontal disease progression in the late 1980’s stated that specific bacteria initiated the disease process by activating host responses, which were protective and destructive. The actual destruction of connective tissue and bone resulted primarily from inflammatory chemical mediators released by immunocompetent cells, such as matrix metalloproteinases, IL-1, and prostaglandins.
Our understanding of the pathogenesis of periodontal diseases was greatly improved by longitudinal studies published during the late 1980’s and 1990’s. Löe et al. (1986) 15 in their classic study of the natural history of periodontitis on tea plantation workers in Sri Lanka found that among individuals with poor oral hygiene and no access to dental care, some developed disease at a rapid rate, whereas others experienced little or no disease. In this study, it was appreciated that some unrecognized environmental factors or some individual differences in susceptibility to the disease were present in the population under study. During the same period, the importance of genetic variations in determining the development and severity of the periodontal disease, with genetic influences accounting for as much as 30% to 60% of the variability in the clinical severity of periodontitis was established 16, 17. Along with this, it was found that smoking 18-21 and diabetes 22-26 were powerful determinants of disease severity. These factors were considered as modifying factors for the final outcome of the disease progression. So, to incorporate all these factors in the pathogenesis of the periodontal disease, non-linear model of disease progression was proposed 27.
Recently, Kornman (2008) 28 put forward a biologic systems model of the pathogenesis of periodontal disease. Although the non-linear model of disease progression still holds well, this model incorporated the role of contributing factors in the pathogenesis of periodontal diseases. According to this model, disease activity depends on …….. Contents available in the book……….. Contents available in the book……….. Contents available in the book……….. Contents available in the book…
Our current understanding of host-microbial interactions
To better understand the host-microbial interactions in periodontal diseases, one should have the basic knowledge of innate and adaptive immune response which has been discussed in the previous chapter. The initiation of inflammatory response occurs with microbial insult derived from microorganisms in the dental plaque. The microbial insult causes initiation of host response that eventually results in tissue destruction.
Microbial insult
As discussed in chapter “Microbiology of periodontal diseases”, the bacterial species in the plaque biofilm may or may not be pathogenic to the host. Various bacterial species which are not associated with the periodontal disease progression are designated as commensals. On the other hand, bacterial species which have been shown to possess virulence factors that cause damage to the host tissues are designated as periodontal pathogens. To produce insult to the host, the microorganism should be able to enter the host, should establish itself in the host, should be able to evade the host response and should be able to produce virulence factors that cause tissue damage. The microorganisms that have been shown to be associated with the periodontal disease progression and virulence factors produced by them have been discussed earlier in chapter 5.
Host response
The host immune response against microbial insult consists of innate and adaptive responses. The first defense against the invading periodontal pathogens and their products is junctional epithelium. The details of the structure of junctional epithelium have been discussed earlier in, “Junctional epithelium”. The cells of the junctional epithelium have fewer desmosomes as compared to the normal epithelial cells, which account for its remarkable permeability 42-44. This permeability is closely related to the ingress of bacteria and their products and outward flow of the gingival fluid and transmigration of neutrophilic granulocytes between the epithelial cells. Because of this reason, the junctional epithelium and the subjacent connective tissue becomes the battlefield for host-microbial interactions.
The cells involved in the first line of defense against many invading microorganisms are macrophages and neutrophils, which are important components of the innate immune response. However, these cells may not always eliminate infectious microorganisms, and some pathogens may not be recognized by them. The adaptive immune response which is specifically directed against these organisms is then generated to eliminate them. Cells involved in the adaptive immune response are lymphocytes. Both innate and adaptive immune responses play a very important role in dealing with these infections.
Initiation of host response by junctional epithelium
The junctional epithelium is constantly exposed to microbial flora which can initiate the inflammatory response. This inflammatory response is primarily mediated by neutrophils, which are the key components of the host defense against bacterial infection. Phagocytic macrophages then play an important role in the recognition of invading microorganism and to initiate the adaptive immune response. As explained in the previous chapter, to better understand the host response against periodontal pathogens, we can divide it into two parts: innate immune response and adaptive immune response. The innate immune response primarily consists of neutrophil’s response, complement system, and Toll-like receptors. The adaptive immune response consists of antigen presentation by antigen presenting cells (APC’s) and generation of T-cell and B-cell response. Let us discuss these responses in periodontal diseases in detail,
Innate immune response
Gingival epithelium provides a physical barrier to infection and has an active role in the innate host defense because the epithelial cells are in constant contact with the bacterial products 45. Along with this, it is now well recognized that epithelium produces a diverse range of antimicrobial peptides which have been found to be belonging to at least four families (α-defensins, β-defensins, cathelicidins, saposins) in humans 46. It has been shown that oral, sulcular, pocket and junctional epithelia of the gingiva are associated with the expression of defensins, more specifically β-defensins (hBD-1, hBD-2, and hBD-3) 45. α and β defensins have an important role in the host immune response. Neutrophils are rich in α-defensins and cathelicidin LL37 (proteolytic peptides). Some investigations have shown that the primary role of β-defensins may be to signal other innate and acquired immune responses, while LL37 and α-defensins may be more important for their antimicrobial properties in the gingival sulcus 47. These peptides are capable of activating the classical complement pathway and appear to upregulate IL-8 production by epithelial cells, which may enhance neutrophil recruitment to the site of infection 48. The polymorphonuclear leukocyte (PMN) appears to play a …….. Contents available in the book……….. Contents available in the book……….. Contents available in the book……….. Contents available in the book…
Complement system
As discussed in “The complement system”, the complement system is a very important component of the innate immune response. It causes the destruction of the microorganisms by the formation of a membrane attack complex. The classical, alternative and lectin pathways of complement system have been well described in detail in the above article.
Role of Toll-like receptors (TLRs) in host-microbial interaction
When microorganisms enter the tissue after penetrating the epithelial barrier, they are encountered by tissue macrophages, mast cells and immature dendritic cells 52. These cells must be able to distinguish between apoptotic particles generated by normal tissue turnover and particles that are indicative of infection. The molecules, mainly responsible for making this pivotal distinction are those of the family of pattern-recognition receptors (PRRs). Mammals have several distinct classes of PRRs including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), Nod-like receptors (NLRs), AIM2-like receptors (ALRs), C-type lectin receptors (CLRs), and intracellular DNA sensors such as cGAS 53, 54. Among these, TLRs were the first to be identified, and are the best characterized. The TLR family comprises 10 members (TLR1-TLR10) in human and 12 (TLR1-TLR9, TLR11-TLR13) in mice. Along with tissue macrophages, mast cells, and immature dendritic cells, these receptors are also expressed on lymphocytes, osteoclast precursors, osteoblasts and stromal and epithelial cells, each of which has different toll-like-receptor expression profiles 55-58. Members of the TLR family are responsible for the recognition of pathogen-associated molecular patterns (PAMPs), expressed by a wide spectrum of infectious agents; and self-derived molecules derived from damaged cells, referred as damage-associated molecular patterns (DAMPs).
TLRs localize to the cell surface or to intracellular compartments such as the ER, endosome, lysosome, or endolysosome, and they recognize distinct or overlapping PAMPs such as lipid, lipoprotein, protein, and nucleic acid. TLRs are largely classified into two subfamilies based on their localization, cell surface TLRs and intracellular TLRs. Cell surface TLRs include TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, whereas intracellular TLRs are localized in the endosome and include TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13 59, 60. The microbial surface-associated components such as lipids, lipoproteins, and proteins are recognized by cell surface TLRs. Bacterial lipopolysaccharides are recognized by TLR4. TLR2 along with TLR1 or TLR6 have the ability to recognize a wide variety of PAMPs including lipoproteins, peptidoglycans, lipoteichoic acids, zymosan, mannan, and tGPI-mucin 59. The bacterial flagellin is recognized by TLR5 53. The intracellular TLRs primarily participate in the recognition of nucleic acids derived from bacteria and viruses, and also recognition of self-nucleic acids in disease conditions such as autoimmunity 61.
The main effect of the stimulation of TLRs is the synthesis and secretion of pro-inflammatory cytokines and lipid mediators, thereby initiating the inflammatory response that recruits both soluble immune components and immune cells from the blood 62.
Structure of Toll-like receptors (TLRs)
Toll receptors were first recognized in Drosophila (fruit-fly). It was found that these were important in the development of innate immunity in adult flies. TLRs are transmembrane proteins expressed by cells of the innate immune system, which are involved in the recognition of invading micro-organisms and initiation of immune and inflammatory responses to destroy the invader 63. TLRs are characterized by an amino-terminal extracellular domain composed of repeated motifs, high in leucine and known as leucine-rich repeats (LRRs), followed by a single transmembrane domain and a globular cytoplasmic domain, called the Toll/IL-1 receptor (TIR) domain, or TIR domain due to its high similarity to that of the IL-1 receptor family. Despite this similarity, the extracellular portions of both types of receptors are structurally unrelated. The IL-1 receptors possess an immunoglobulin-like domain, whereas TLRs bear leucine-rich repeats (LRRs) in the extracellular domain.
Signaling pathways
TLR signaling pathways basically consist of,
- MyD88-dependent pathway (common to all TLRs),
- MyD88-independent pathway (peculiar to theTLR3 and TLR4).
MyD88 (Myeloid differentiation primary-response protein-88) is an adapter molecule that functions to recruit IRAKs (IL-1 receptor-associated protein kinases) to the IL-1 or TLR4 receptor complexes after IL-1 or LPS stimulation, respectively.
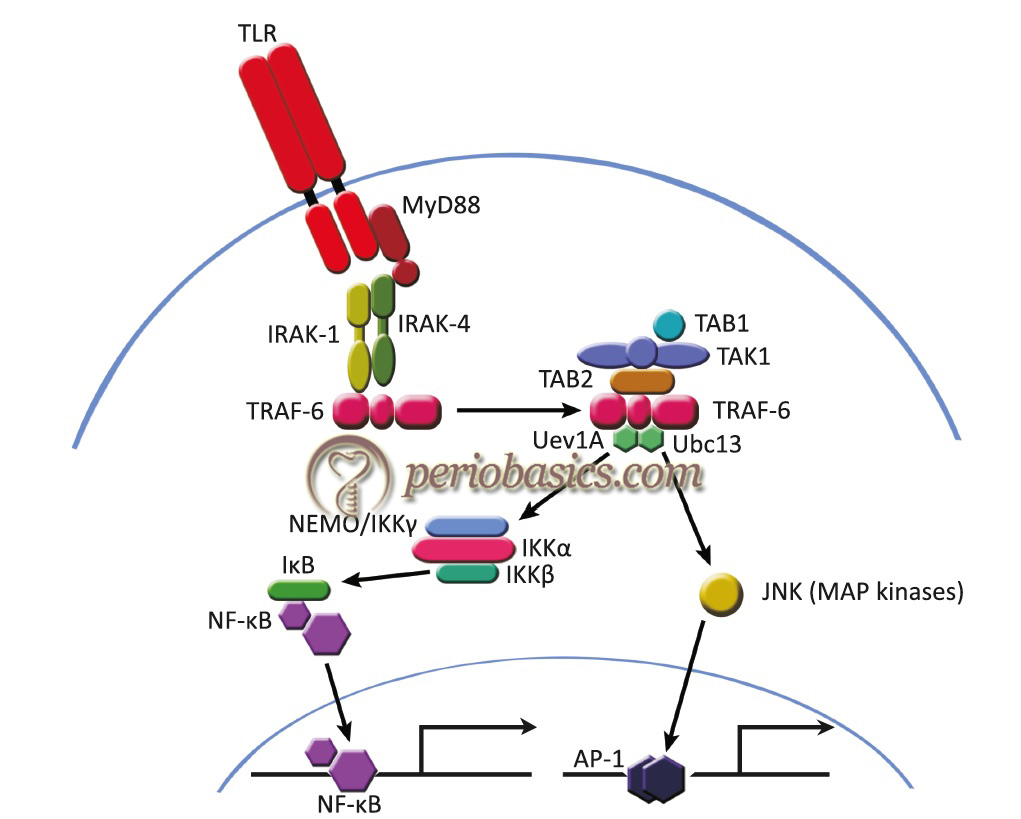
MyD88-dependent pathway
The main steps involved in the MyD88-dependent pathway are as follows,
- Upon stimulation, MyD88 recruits IL-1 receptor-associated kinase (IRAK) to TLRs.
- IRAK is activated by phosphorylation.
- IRAK then associates with TRAF6.
- Then TRAF6 interacts with TAK1, TAB1, and TAB2.
- The complex of TRAF6, TAK1, TAB1, and TAB2 further forms a larger complex with Ubc13 and Uev1A, which induces the activation of TAK1.
- Activated TAK1 phosphorylates the IKK complex, consisting of IKKα, IKKβ, and NEMO/IKKγ, and MAP kinases, such as JNK, and thereby induces the activation of the transcription factors NF-κB and AP-1, respectively.
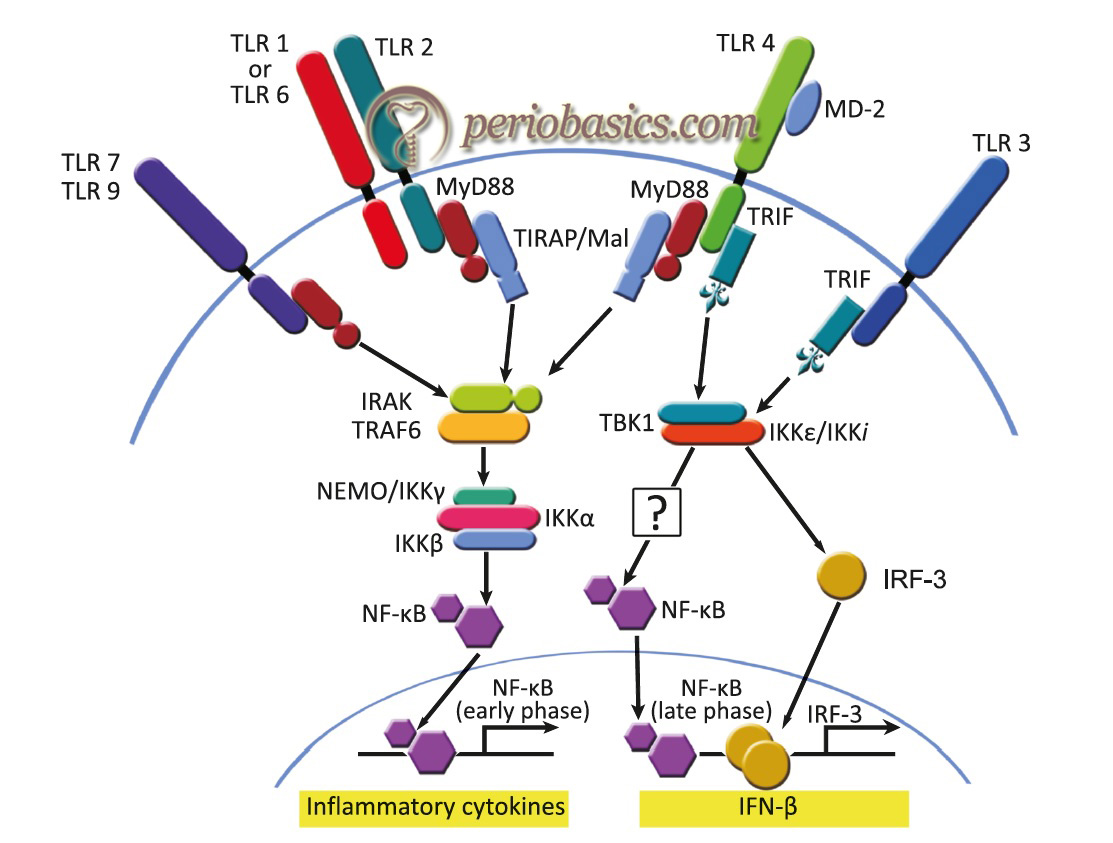
MyD88-independent pathway
MyD88-deficient mice have been generated and found to be completely defective in their responses to IL-1 and IL-1-related cytokine, IL-18 65. The response to LPS was also shown to be abolished 66. Although MyD88 plays a critical role in TLR signaling, there is also present a MyD88-independent pathway. MyD88-independent signaling pathway is shared by TLR3 and TLR4 cascades. TIR-domain-containing adapter-inducing interferon-beta (TRIF or TICAM1) is a key adapter molecule in transducing signals from TLR3 and TLR4 in a MyD88-independent manner. In a step by step manner, it is as follows,
- TRIF is recruited to ligand-stimulated TLR3 or 4 complex via its TIR domain.
- The activation results in recruitment of IKKε/TBK1, phosphorylation of IRF3, and expression of interferon-β.
- The result is the production of type 1 interferons (IFNs), pro-inflammatory cytokines and induction of programmed cell death.
- Although MyD88 is reported to be involved in TLR3 signaling 67, TLR3 does not appear to use the MyD88-dependent pathway to any significant extent, because the response to poly (I-C) (polyinosine-polycytidylic acid) was not impaired in MyD88-deficient mice 68.
Result of TLR activation
As already stated, TLR activation results in the production of various pro-inflammatory cytokines and chemokines. The interaction of TLR4 on dendritic cell and LPS results in the production of pro-inflammatory cytokines, such as IL-12, and the interaction of TLR3 with LPS results in the production of type-I interferon. It has been demonstrated that the most predominant TLR’s in the periodontal tissues are TLR2 and TLR4 69. It must be noted that the TLR response may vary…….. Contents available in the book……….. Contents available in the book……….. Contents available in the book……….. Contents available in the book…
Adaptive immune response
The adaptive/acquired immune response is activated when the epithelial barrier, with its antimicrobial peptides and other components of innate systems, is breached. The pathogenic species present in the subgingival biofilm evade the anti-bacterial host defense mechanisms by releasing an array of virulence factors, which causes damage to the host tissue by immune/inflammatory interactions, which typically consist of neutrophils, monocytes/macrophages, dendritic cells (DCs), T-cells, and predominantly IgG-producing plasma cells. The majority of virulence factors include enzymes and endotoxins.
T-cell activation in the adaptive immune response
The T-cell adaptive immune response is activated by processing and presentation of bacterial antigens by lymphocytes, macrophages, and dendritic cells. After phagocytosis of bacteria, its recognizable surface antigens are presented on the surface of APC’s.
Antigen-presenting cells (APC’s)
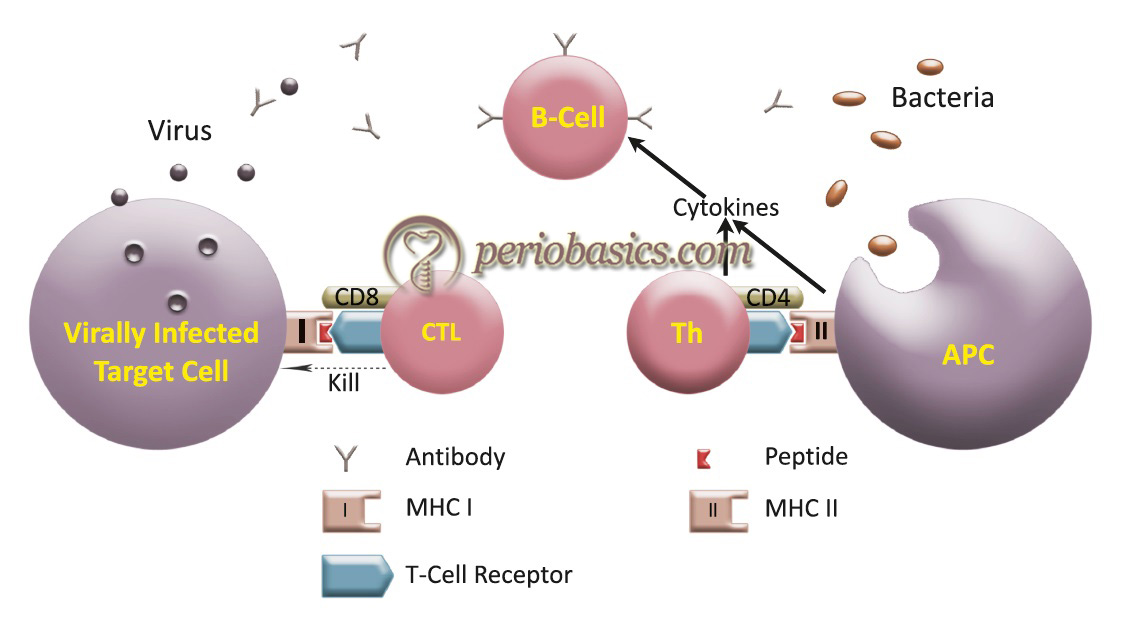
The APC’s have the antigenic peptide with major histocompatibility complex (MHC) molecule located at their surface. Cytotoxic T-lymphocytes (CTL) expressing the CD8 co-receptor recognize the peptide bound to MHC Class I molecules, whereas helper T-cells (Th) expressing the CD4 co-receptor does so with a peptide associated with MHC Class II molecules. Co-stimulatory molecules such as B7-1 (CD80) and B7-2 (CD86) are also present on APC’s that interact with CD28 on T-cells. Adhesion molecules such as ICAM-1 present on APC’s are involved in the formation of strong immunological synapses to facilitate the proper activation of T-cells.
Interactions between APC’s and T-cells
To understand the interaction between T-cells and antigen-presenting cells, it is important to understand the receptors present on these cells and their interactions. There is a series of intracellular signaling cascade that is activated when a receptor is activated, which ultimately leads to the synthesis and secretion of biochemical mediators like cytokines, etc. The interface between lymphocytes and targets is termed ‘immunological synapse’ (IS).
T-cell receptor (TCR)
One of the initial steps in the generation of the immune response is the recognition by T-lymphocytes of peptide fragments (antigens) derived from foreign pathogens that are presented on the surface of APC’s. This event is mediated by the TCR, which transduces these extracellular signals by initiating a wide array of intracellular signaling pathways. The TCR is a complex of integral membrane proteins that participate in the activation of T-cells in response to the presentation of antigen by APC’s. MHC molecules on APC’s that present antigen peptides to TCR complexes trigger TCR and induce a series of intracellular signaling cascades. Engagement of the TCR with APC’s initiates positive (signal-enhancing) and negative (signal-attenuating) cascades that ultimately result in cellular proliferation, differentiation, cytokine production, and/or activation-induced cell death.
TCR is composed of six different chains that form the TCR heterodimer responsible for ligand recognition. CD3 molecules (CD3-g, CD3-d, CD3-e, and CD3- z), which are assembled together with the TCR heterodimer, possess a characteristic sequence motif for tyrosine phosphorylation, known as ITAMs (Immunoreceptor Tyrosine-based Activation Motifs). One of the initial steps following TCR activation is the activation of Src family tyrosine kinases (p56lck) that, in turn, phosphorylate phospholipase Cg1 (PLC g1). Activation of PLC g1 leads to hydrolysis of phosphatidylinositol 4, 5-bisphosphate (PIP2), generating diacylglycerol (DAG) and inositol trisphosphate (IP3). DAG activates protein kinase C (PKC) that, in turn, phosphorylates Ras, a GTPase that activates Raf, leading to the recruitment of the MAP kinase cascade. IP3 releases calcium from its intracellular stores in the endoplasmic reticulum (ER).
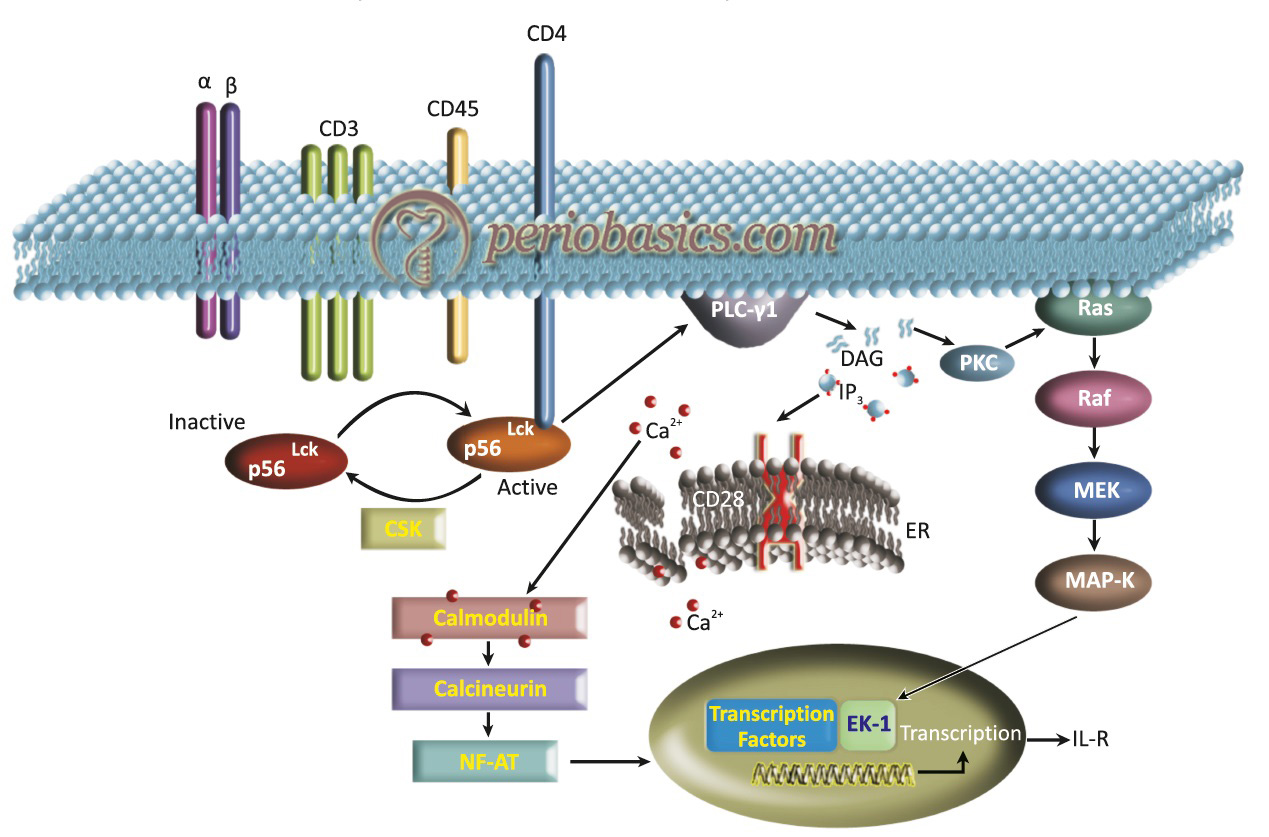
The Ca2+ binds to calmodulin that, in turn, activates calcineurin, a Ca2+/calmodulin dependent protein phosphatase. NFAT, a transcriptional regulator of IL-2 gene expression, is a direct target of calcineurin. Calcineurin dephosphorylates the cytosolic component of NFAT, NFATc, which migrates to the nucleus and induces transcription of the IL-2 gene. Adhesion molecules like LFA-I interact with ICAM-I on APC’s and aid in the formation of the immunological synapse.
To mount an immunological response, the T-cell needs to receive a second signal from an antigen-presenting cell in the form of a co-stimulatory molecule. Co-stimulatory molecules act through different TCRs, such as the CD28 and TNFR (tumor necrosis factor receptor) families, producing a second signal that induces T-cell activation and proliferation. The CD28 and TNFR co-stimulatory receptors, possibly exert their effect through promoting more efficient signaling by concentrating the kinases and substrates that are required to initiate a signal whose affinity has been shown to increase upon stimulation.
Steps in the activation of T-cells
- Antigen which is phagocytosed by a macrophage is cleaved into polypeptides which are then transported to the surface for presentation to T-cells (only foreign particles consisting of proteins are activated and processed, no other molecules like fatty acids etc. are presented).
- The APC complex consists of both antigen and MHC. If MHC Class-II is associated with presenting cells, CD4 or helper T-cells are activated and if MHC Class-I associated with presenting cells, CD8 or cytotoxic T-cells are activated. MHC complex is encoded by a group of genes, so different polypeptides are presented on the surface of the MHC which is responsible for its diversity of antigen presentation. MHC presents only proteins which may be derived from foreign or self-proteins. It depends on the selection of T-cells in the thymus.
- Macrophage which is attached to the antigen, produces IL-1 which activates CD4 cells.
- CD4 interacts with MHC Class-II on APC surface. This union is stabilized by other proteins LFA-1 on T-cells and ICAM 1 on APC.
- A co-stimulatory signal is formed by B7 protein on APC and CD28 on CD4 cells which result in the secretion of IL-2 by the helper T-cells and it is this step that is useful in the execution of all the functions i.e., regulator, effector, and memory functions. Production of IL-2 is the most crucial step in T-cell activation. If this co-stimulatory signal is not formed, anergy takes place.

Cell-mediated immune response in periodontal diseases
T-lymphocyte response to antigenic challenges is called as a cell-mediated immune response. T-lymphocytes can be functionally divided into CD4 (helper T- lymphocytes) cells and CD8 (cytotoxic T-lymphocytes) cells by the type of antigen receptors and a small number of accessory markers on their cell surface. Before we go into details of the cell-mediated immune response in periodontal diseases, let’s first try to understand helper T-cells.
Helper T-cells (Th cells)
Helper T-cells are pivotal for the development of protective immune responses. The Th cells may belong to different cell lineages as they emerge from the thymus such as “natural” regulatory T (nTreg) cells and natural killer cells (NK-cells); or they may demonstrate alternative patterns of differentiation of naïve CD4 T-cells. It is important to understand here about naïve CD4 T-cell (Thn cells). Thn cell is a T-cell that has differentiated in bone marrow, and successfully undergone the positive and negative processes of central selection in the thymus; however, unlike activated or memory T-cells, has not encountered its cognate antigen within the periphery. When Thn cells are exposed to antigen by APC’s, it results in the differentiation of Thn cells into…….. Contents available in the book……….. Contents available in the book……….. Contents available in the book……….. Contents available in the book…
Let us now discuss these four types of Th-cell populations,
Th1 cell
These cells mediate immune response against intracellular pathogens 88, 89. These are the main cells in generating an immune response against mycobacterial infections. The Thn cells give rise to Th1 cells under the influence of IFN-γ and IL-12. IL-12 causes Th1 differentiation and blocks Th2 cell production, while IL-4 causes Th2 differentiation and antagonizes Th1 development. IL-18 also induces Th1 differentiation. Their main secretions of Th1 cells are IFN-γ, lymphotoxin-α (LT-α), and IL-2. IFN-γ is an important stimulator of macrophages for increasing their microbicidal activity. IL-2 production is important for CD4 T-cell memory. Also, IL-2 is an important cytokine involved in stimulation of CD8 cells during their priming phase and results in the formation of CD8 memory cells.
Th2 cells
These cells are involved in the generation of immune response against extracellular parasites including helminths. The Th2 cells get differentiated from Thn cells under the influence of IL-2 and IL-4. Th2 cells influence B-cell activation, proliferation and immunoglobulin production. Their secretions are IL-4, IL-5, IL-9, IL-10, IL-13, IL-25, and amphiregulin. IL-4 stimulates B-cell growth and heavy chain switch from IgM to IgG, IgE, and IgA and stimulates high-affinity antibody synthesis. These cytokines also induce proliferation of basophils/mast cells by IL-4 and the proliferation and differentiation of eosinophils by IL-5. Thus, Th2 cells are important in the induction and persistence of asthma and other allergic diseases. IL-10, produced by Th2 cells, suppresses Th1 cell proliferation 90. Amphiregulin is a member of the epidermal growth factor (EGF) family and it induces epithelial cell proliferation. IL-13 is involved in the induction of airway hypersensitivity. IL-25 functions as an initiation factor as well as an amplification factor for Th2 responses.

Th17 cells
The Th17 cells represent a third effector arm of CD4 T-cells and complement the function of the Th1 and Th2 cell lineages. These play a critical role in the induction of tissue inflammation and tissue destruction that are hallmarks of many immune-inflammatory diseases. These cells mediate immune responses against extracellular bacteria and fungi 91. These get differentiated from Thn cells under the influence of TGF-β, IL-6, IL-21, and IL-23. Th17 cells produce IL-17a, IL-17f, IL-21, and IL-22.
Treg cells
Treg cells are a heterogeneous T-cell subpopulation that regulates the immune system in various ways. They play a critical role in maintaining self-tolerance as well as in regulating immune responses. In humans, Treg cells contribute to 5%-10% of peripheral CD4 T-cells and constituently express several suppressing mediators, such as CD25 (α chain IL-2 receptor), glucocorticoid-induced TNF-R (GITR), cytotoxic T-lymphocyte Ag-4 (CTLA-4), CD103 and the transcription factor Foxp3 92. These cells get differentiated from Thn cells under the influence of TGF-β and IL-2. TGF-β produced by Treg cells may also result in the induction of iTreg cells from naïve CD4 T-cells.
Th0, Th1 and Th2 cell-mediated immune response in periodontal diseases
The immune response to infection is regulated by the balance between Th1 and Th2 cytokines 93. Studies have been done to find out Th1/Th2 immune response in periodontal disease. Investigations indicated a dominance of the Th1 response over the Th2 response, with other studies showing a predominance of Th0 cells in periodontitis 94, 95. Various studies have reported discrepancies with regard to the predominance of Th1 or Th2 response or the involvement of both Th1 and Th2 cells in the diseased tissue 96-99.
Role of Th17 cells in periodontal diseases
A distinct type of helper T-cell lineage, Th17 has been recently identified. This population secretes several pro-inflammatory cytokines, including the novel cytokine IL-17, and, hence, has been termed ”Th17”. It expands in the presence of IL-23 100. IL-23 is recently discovered cytokine which belongs to the IL-12 family 101. The Th17 cell lineage is thought to play an important role in the pathogenesis of cell-mediated tissue damage caused either by autoimmunity or immune responses against microbial infection 102. Th17 cells have been proposed to exert both pro- and anti-inflammatory functions. Further, these cells have also been shown to express higher levels of RANKL than Th1 cells 103. The exact role of these cells in periodontal diseases still needs to be investigated.
Role of Regulatory T-cells in periodontal diseases
The evidence regarding the exact role played by Treg cell in mediating the periodontal disease continues to be controversial. Studies demonstrated that periodontitis patients showed an increased percentage of Treg cells in the gingival connective tissue as compared to gingivitis patients. It was concluded that Tregs levels were upregulated in chronic periodontitis lesions as protection against self-antigens, such as collagen Type-I 104, 105. Another study showed that Treg cells downregulated RANKL expression in periodontal disease. However, their capacity to provide an immunoregulatory function in periodontal disease has been questioned 106. One study suggested that Treg cells may not have a regulatory function due to the lack of CTLA-4 expression that is required for cell-cell contact inhibition of T-cell proliferation 107.
Hence, further investigations are required to find out the exact role of regulatory T-cells in host-microbial interactions in periodontal diseases.
B-cell activation in adaptive immune response
The B-cell mediated or the humoral immune response is mediated by the production of antibodies by cells of B-lymphocyte lineage. B-cells express a unique B-cell receptor (BCR) on their surface that recognizes and binds to only one particular antigen. The primary differences between B-cell and T-cell mediated immune response is in the mechanism by which they recognize the antigen. As already discussed, T-cell recognizes the antigen through MHC interactions, once it is presented by the APC’s. The B-cell recognizes antigens in their native form. After a B-cell recognizes the antigen and receives appropriate signals from helper T-cells, it further differentiates into an effector cell, known as a plasma cell. The plasma cells produce antibodies against that antigen. These cells are short-lived cells (2-3 days). However, around 10% of plasma cells become long-lived and are referred to as antigen-specific memory B-cells. The antibodies, thus produced, bind to the antigen, making it an easier target for phagocytes. The B-cell response may be T-cell dependent or T-cell independent, but it is primarily regulated by T-cells.
T-cell independent activation of B-cells
T-cell independent response occurs by the cross-linking of the IgM by antigen receptors on the B-cell, responding with IgM synthesis in the absence of T-cell help.
T-cell dependent activation of B-cells
When a pathogen is ingested by an APC such as a macrophage or dendritic cell, the pathogen’s proteins are then digested to peptides and attached to a Class II MHC protein. This complex is then moved to the outside of the cell membrane. The APC, then interacts with the helper T-cell (which recognizes MHC II). This activation leads to the secretion of cytokines by helper T-cell, which causes B-cell proliferation and maturation. Activated B-cells subsequently produce antibodies, which assist in the inhibition of pathogens.
Humoral immune response in the periodontal diseases
Although the precise role of humoral immunity in the periodontal disease progression has not been fully elucidated, however, it has been suggested that the humoral immune response has a protective role in the pathogenesis of periodontal disease 108. B-lymphocytes contribute to immunity in multiple ways, including the production of antibodies, presentation of antigen to T-cells, organogenesis of secondary lymphoid organs and secretion of cytokines 109. The antibodies protect bacterial colonization primarily by two mechanisms …….. Contents available in the book……….. Contents available in the book……….. Contents available in the book……….. Contents available in the book…
After encountering the invading pathogens, in particular, extracellular microorganisms, the antigen (Ag)-specific naïve B-cells undergo affinity maturation via clonal selection, somatic hypermutation, and Ig receptor editing 114. The B-cells bearing the higher or highest affinity for Ag are rescued from apoptosis and subsequently, these B-cells with mutated B-cell receptor genes, located in the germinal centers of regional lymph nodes, undergo further isotype-switching and differentiate into effector or memory B-cells 115. Certain cytokines such as interferon-γ (IFN-γ), IL-4, IL-5, IL-10, IL-12, transforming growth factor-β (TGF-β), anti-CD40-Ligand/CD40, and bacterial LPS are potent stimulators of antibody class-switching 116, 117.
Immunoglobulins arriving at the periodontal lesion are from both systemic and local tissue sources. The salivary IgA is involved in trapping of the antigen in a mucin layer with the subsequent disposal of the antigen 118. The local production of IgG is protective in nature and is found to be increased in inflamed gingival and periodontal tissues 119.
Review of literature on humoral response in periodontal diseases
Many studies have examined the antibody response in various periodontal diseases. In a study, Ranney and co-workers (1982) 124 examined the presence of antibody response against A. actinomycetemcomitans in 57 juvenile periodontitis patients. The results demonstrated that 31 out of 57 patients were positive for the antibodies and antibody response was significantly higher in localized juvenile periodontitis and severe periodontitis patients as compared to healthy patients. A more extensive study was done by Gunsolley et al. (1987) 125, where they examined the antibody response against 25 Gram-negative, Gram-positive, and spirochetal micro-organisms and their relationship with the attachment loss. The results of the study demonstrated that there was a positive correlation between antibody response and percentage of teeth with 2-mm, 5-mm attachment loss, and the mean attachment loss. Antibody response against 11 bacterial species demonstrated a positive correlation with the severity of the disease.
Many studies have suggested that patients with localized aggressive periodontitis have elevated antibody levels and they also demonstrate high serum concentrations of IgG2 126-128. Studies have also been done on the genetic association of high serum levels of IgG2 in aggressive periodontitis patients. They have demonstrated the heritability of serum IgG2 concentrations in families with aggressive periodontitis 129, 130. In a study, Tew et al. (1985) 131 studied the serum IgG levels in 52 severe periodontitis, 47 localized juvenile periodontitis, and 52 healthy patients. The authors examined the antibody response against Eubacterium species, Lactobacillus minutus, and Peptostreptococcus micros using RIA. These bacteria were selected because of their predominance in cultivable flora from the subgingival plaque of the patients with severe periodontitis. The results of the study demonstrated that severe periodontitis patients were seropositive for E. brachy and P. micros, but not the juvenile periodontitis and healthy patients. The juvenile periodontitis patients had elevated antibody levels against E. timidum than observed in healthy patients. It was concluded by the authors that many micro-organisms in the subgingival flora elicit serum antibody; however, some bacteria that are present in high numbers do not elicit significant antibody responses.
Know More…
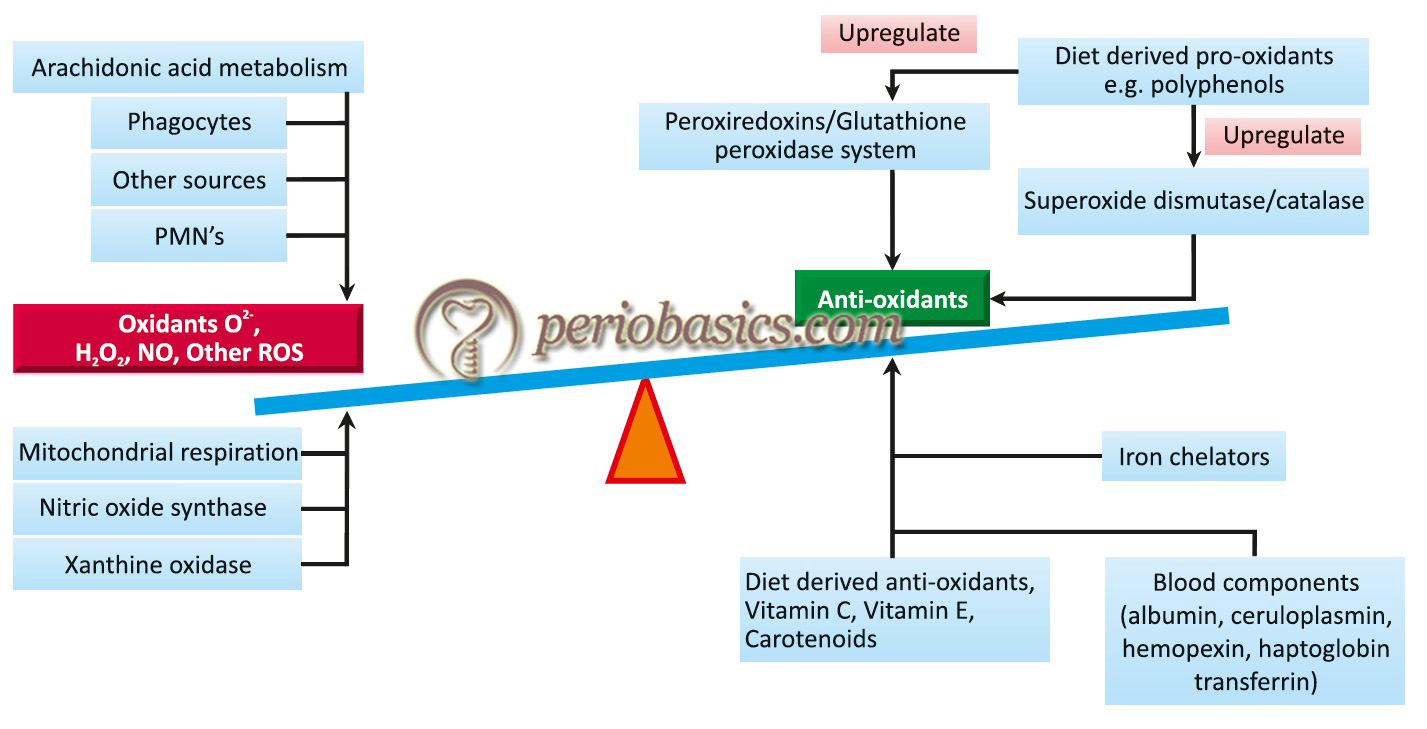
Oxidative stress:
One important aspect of host-microbial interactions is the generation of oxidative stress. Oxidative stress is altered physiological balance between oxidants and antioxidants in favor of the former leading to lesions in the body. The resultant lesion is known as ‘oxidative damage’. During health, a dynamic balance is maintained between the oxidants and anti-oxidants in our body. However, this balance may be disturbed as a result of an increase in the concentration of reactive oxygen species (ROS) and/or decreased concentration of the anti-oxidants. As discussed earlier, during host-microbial interactions, the stimulated PMN’s and other immune cells release an increased amount of ROS, culminating with oxidative lesions in the gingival tissue, periodontal ligament, and alveolar bone.
These reactive species initially cause depolymerization of extracellular matrix components, followed by lipid peroxidation, degradation of enzymes and induction of cytokine synthesis. Along with this, the ROS can pass through the nuclear membrane and cause oxidative damage to the DNA. The most studied oxidative DNA damage in aggressive periodontitis is the formation of 8-hydroxydeoxyguano-sine (8-OHdG) 132, which is a change that occurs in about one in 105 of the guanosine bases in the normal human cells 133, 134. In lesions that result from the oxidative damage to the nuclear and mitochondrial DNA, 8-OHdG and 8-oxo-7,8-dihydro-2′- deoxyguanosine (8-oxodG) are predominantly found, therefore they may act as biomarkers for oxidative stress.
Conclusion
The understanding of the host-microbial interactions is the cornerstone of our overall understanding of the etiopathogenesis of periodontal diseases. There are multiple mechanisms by which our immune system responds to the bacterial insult. In the above discussion, we discussed various aspects of host-microbial interactions. In the next articles, we shall read about the “Models of periodontal disease progression” and “Microbiology of periodontal diseases”.
References
References are available in the hard-copy of the website.