Introduction to desquamative gingivitis
Desquamative gingivitis (DG) is the clinical term given to the gingival manifestation of mucocutaneous diseases. This gingival condition is characterized by intense erythema, sloughing off or ulceration that is commonly associated with some kind of systemic autoimmune disease. This condition is non-plaque associated and may be localized to the marginal gingiva, the attached gingiva or both. It is not a specific diagnosis, but rather a sign of the underlying disease. It is more common in middle-aged to elderly females. As compared to plaque-induced gingivitis, which commonly involves marginal gingiva, desquamative gingivitis mostly affects buccal/labial gingiva, frequently sparing the marginal gingiva but can involve the whole thickness of the attached gingiva. Most importantly, its clinical appearance is not significantly altered by traditional oral hygiene measures or conventional periodontal therapy alone 1, 2.

Historical aspect
Chronic desquamative gingivitis was first described by Tomes and Tomes in 1894 3. The term “Desquamative gingivitis” was coined by Prinz in 1932. It is a descriptive term synonymous with the presence of erythema, desquamation, erosion, and blistering of the attached and marginal gingiva 4. Schour and Massler (1947) 5 used the term “Gingivosis” to describe this condition in a group of malnourished Italian children. Initially, it was thought to be a specific degenerative disease of gingival tissue with unknown etiology. In 1960 McCarthy and colleagues suggested that desquamative gingivitis was not a specific entity, but a gingival response associated with a variety of conditions 6. This was further concluded by Glickman and Smulow in 1964 7. Presently, many systemic autoimmune disorders and other conditions have been described to have desquamative gingival lesions.
Etiopathogenesis of desquamative gingivitis
To understand the pathogenesis of desquamative gingivitis, first, we must know the basic organization of the oral mucosa. Oral mucosa is composed of a stratified epithelial layer and an underlying connective tissue. In the oral cavity, keratinized and non-keratinized oral mucosa is present. In the hard palate and marginal gingiva, the epithelium of the mucosa is ortho- or para-keratinized to resist the mechanical trauma caused by masticatory forces. The dorsum of the tongue and vermilion border of the lips consist of both keratinized and non-keratinized epithelium. The epithelium is non-keratinized in the buccal mucosa, alveolar mucosa, the floor of the mouth, ventral tongue, soft palate, and lips 8-10.
There are two distinct layers in the oral mucosa: the stratified squamous cell epithelium and the lamina propria. These two layers are separated by a basement membrane. The basement membrane consists of collagen Type IV, laminin, and heparin-proteoglycans. It connects the differentially keratinized epithelial regions with the underlying submucosal connective tissue. The lamina propria consists of a layer of loose fatty or glandular connective tissue containing the major blood vessels and nerves, which separates the oral mucosa from the underlying bone or muscle 11, 12. The epithelial layer is stratified into four layers: stratum basale, stratum spinosum, stratum granulosum, and stratum corneum. The structural organization of these layers has been discussed in detail in “Periodontium in health”.
Know More…
Components of basement membrane zone at electron microscopic level:
Often the terms “basement membrane” and “basement membrane zone” are used interchangeably. At electron microscopic level, the basement membrane zone is divided into four distinct layers. These four distinct layers are the: (1) the basal keratinocyte layer, (2) the lamina lucida, (3) the lamina densa, and (4) the sublamina densa (superficial papillary dermis). Within basal keratinocytes, there are keratin intermediate filaments, hemidesmo-somes, composed of bullous pemphigoid antigen 1 (BP1) & 2 (BP2), plectin, α6β4 integrin, α3β1 integrin, collagen Type XVII, and CD151. Within the lamina lucida are anchoring filaments, composed predominantly of Laminin 332. Within the lamina densa are collagen Type IV, Laminins 332, 311, and 511, nidogen and perlecan. Within the sublamina densa are anchoring fibrils, composed of collagen Type VII.
Desmosomes and hemidesmosomes are involved in maintaining the epithelial-intercellular and keratinocytes-basement membrane adhesion, respectively. Desmosomes which are responsible for intercellular attachment consists of two principal groups of proteins: desmosomal cadherins, desmogleins (Dsg) and desmocollins, and a large group of plaque-associated proteins. In addition, epithelial cells are also connected by the gap and tight junctions. Narrow intercellular spaces contain proteoglycans and glycoproteins 13, 14.
The cadherins are a major class of membrane proteins with prominent roles in cell adhesion and regulation of tissue organization and morphogenesis. They are composed of an extracellular domain, involved in calcium-dependent binding to adjacent cells, a transmembrane and an intracellular domain that binds to catenins and thence to actin 15. Desmogleins are glycoproteins belonging to the cadherin-supergene family. They link to cytokeratins via desmoplakins and plakoglobin. There are four desmoglein isoforms, designated as desmoglein 1-4. Expression of desmoglein 1 and 3 is restricted to stratified squamous epithelia. In skin, both desmoglein 1 and 3 are expressed, but in the oral epithelium desmoglein 3 is preferentially expressed. Hemidesmosomes are involved in cell and basement membrane contact, linking the keratinocyte cytoskeletons to the lamina lucida. The hemidesmosome associated proteins are bullous pemphigoid antigen 1 (BP1), bullous pemphigoid antigen 2 (BP2), α6β4 integrin, laminin 5, laminin 6, uncein, collagen Type VII and Type IV.

These proteins act as cementing material for the maintenance of intercellular and cell-basement membrane integrity. The autoimmune response against any of these epithelial proteins results in the loss of cell to cell adhesion or loss of cell basement membrane adhesion, leading to vesiculation. In various autoimmune dermatological disorders, antibodies are formed against one or more of these proteins. Following table enumerates various antigens which are targeted by antibodies in vesiculobullous lesions, most of which involve the oral cavity.
| Autoimmune dermatological disorder | Antigen |
|---|---|
| Pemphigus vulgaris | Desmoglein 1 and 3 |
| Paraneoplastic pemphigus | Desmoglein 1 and 3, plakin proteins |
| Pemphigus foliaceous | Desmoglein 1 |
| IgA pemphigus | Dsg3, desmocollin 1 and 2 |
| Pemphigus herpetiformis | Desmoglein 1 |
| Cicatricial pemphigoid | BPAg1 and BPAg2/ laminin V/ laminin VI/ α6-integrin subunit/ β4-integrin subunit/ collagen VII |
| Bullous pemphigoid | BP 180 and 230 |
| Epidermolysis bullosa acquisita | Type VII collagen |
| Epidermolysis bullosa simplex | Keratin 5 and 14 |
| Epidermolysis bullosa junctional | Laminin 5 and type XVII collagen |
| Epidermolysis bullosa dystrophic | Type VII collagen |
| Erythema multiforme | Desmoplakins |
| Dermatitis herpetiform | Tissue transglutaminase |
Classification of desquamative gingivitis
Desquamative gingivitis is an oral manifestation of many systemic conditions. Based on the etiologic considerations, together with histologic and immunologic findings, diseases and conditions which may have desquamative gingivitis as one of their clinical features are classified as,
A. Dermatological diseases,
Lichen planus.
Pemphigus.
Bullous pemphigoid.
Cicatricial pemphigoid.
Linear IgA disease.
Epidermolysis bullosa acquisita.
Dermatitis herpetiformis.
Erythema multiforme.
Lupus Erythematosus.
Contact stomatitis.
B. Drug-induced oral reactions.
C. Endocrine disturbances.
D. Aging.
E. Chronic infections.
F. Other causes.
Diagnosis of desquamative gingivitis
A detailed case history of the patient is recorded along with complete intraoral and extra-oral examination. A detailed family history is also recorded because some diseases such as systemic lupus erythematosus are inherited disorders. The clinical appearance of the oral and extraoral lesions is recorded, if they are present. The oral lesions may be present in the form of white striae, papules, vesicles, and ulcers. Sometimes, the patient is asymptomatic but may complain of soreness, especially when eating spicy or acidic food, and of bleeding and discomfort with toothbrushing. The patient may also give a history of blood filled oral blisters or extensive oral ulceration. The patient is asked relevant questions regarding ocular, nasal and throat symptoms, skin or genital lesions and details of current medication, as well as changes in medication prior to the onset of symptoms.
| Disease | Immunofluorescence findings |
|---|---|
| Pemphigus vulgaris | IgG, IgA, and IgM in intraepithelial layer |
| Bullous pemphigoid | IgG and C3 in epidermal basal layer |
| Mucous membrane pemphigoid | IgG and C3 in epidermal basal layer |
| Paraneoplastic pemphigus | IgG and complement in intraepithelial layer and basement membrane |
| Linear IgA disease | Linear IgA along the epidermal-dermal border |
| Dermatitis herpetiformis | Granular IgA and complement in the tips of the papillary dermis |
| Chronic ulcerative stomatitis with SES (stratified epithelial-specific) ANA (antinuclear antibody) | Speckled ANA in lower third of epidermis |
| Lupus erythematosus | IgG and IgM in basement membrane |
| Erosive lichen planus | IgM in colloid bodies in papillary dermis |
| Erythema multiforme | IgM and C3 in blood vessel walls |
The buccal and labial gingiva is more commonly affected by DG lesions as compared to lingual and palatal gingiva. In some cases, there may be generalized gingival involvement. Nikolsky’s sign often shows a positive reaction in patients with DG. A positive Nikolsky’s sign, where a blister forms or spreads, or the surface epithelium “floats away” when lateral pressure is applied to the mucosa, may indicate vesiculobullous disorders. This sign can be induced in clinically healthy gingival tissue immediately adjacent to the lesional tissue by using a wooden tongue depressor or a pencil eraser to apply gentle friction on the area. An incisional biopsy that includes the lesion and perilesional tissue for histology and immunofluorescence is the recommended way to make a definitive diagnosis.
Dermatological diseases associated with desquamative gingivitis
Lichen planus
Lichen planus (LP) is a relatively common, chronic mucocutaneous disease that often affects the oral mucosa. It is a chronic inflammatory disorder which affects stratified squamous epithelia and is potentially premalignant. This disorder usually develops between the age of 40-70 years, and women are affected more often than men 16. Initially, LP was recognized as a skin disease; however, it was seen that it had frequent oral involvement. It has been observed that more than one-third of the patients with LP of skin had oral lesions, whereas only 12-14% of patients with oral LP had skin lesions 17. Six P’s (planar, purple, polygonal, pruritic, papules, and plaques) are used to describe the skin lesions of LP. The skin lesions are frequently found on the flexor surfaces of the body. Less common sites for skin lesions are genital, ocular, bladder, nasal, laryngeal and auricular mucosae.
Oral lesions:
Oral lichen planus (OLP) has a bilateral distribution that typically affects the buccal mucosa, dorsum and ventral surfaces of the tongue and/or gingiva. OLP is usually asymptomatic, although when there are areas of erosion or ulceration, the patient may have variable amounts of discomfort, being particularly troublesome when eating spicy or acidic type foods. Andreason (1968) 18 classified OLP lesions into six different forms,
Reticular: The reticular form is the most common form of oral lichen planus which appears as a network of connecting and overlapping white lines or plaques. These lesions are often painless, although patients may complain of a slight roughness or dryness of the affected mucosal surfaces.
Papular: In this form of OLP, small white raised areas approximately 1-2 mm in diameter are seen, which are usually found on the buccal mucosa and dorsum of the tongue, although may also present on other mucosal surfaces.
Plaque-like: Here, areas of homogenous whiteness are seen on the oral mucosa. This form of OLP is typically found on the buccal mucosa or dorsum of the tongue and may be more prevalent amongst those who are smokers.
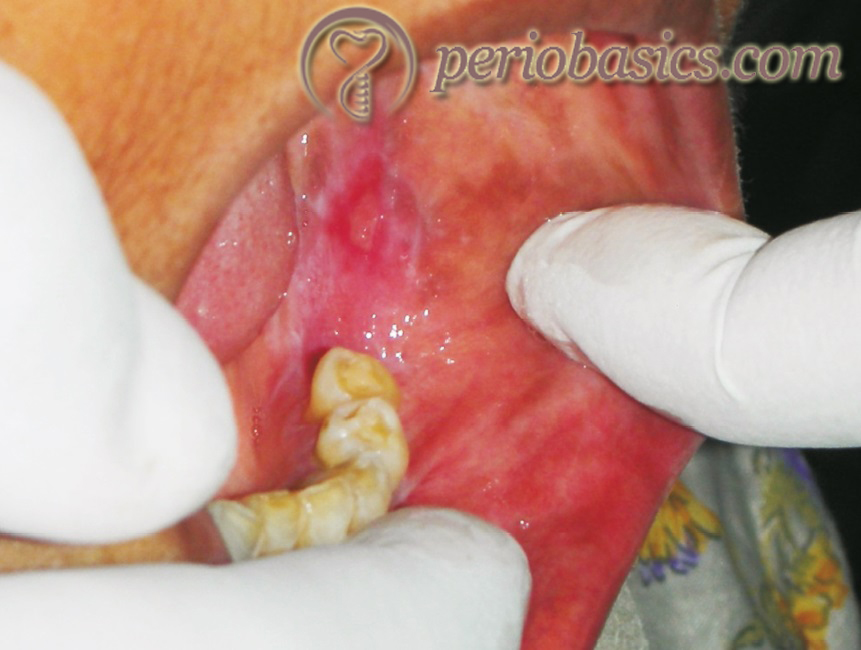
Atrophic: It is also known as erosive oral lichen planus. In this form, there are areas of redness within the aforementioned white patches. The patient complains of oral soreness (Figure 26.4).
Ulcerative: In this case, there are areas of ulceration present within the white areas. The patient complains of continued soreness, this being particularly severe with spicy or acidic foods.
Bullous: This form of OLP is rarely found and is characterized by the presence of small vesicles or blisters (bullae) within the white patches.

Etiopathogenesis of oral lichen planus:
The etiopathogenesis of OLP has been a matter of controversy. However, the majority of evidence supports role of immune dysregulation in the pathogenesis 19-21. Various mechanisms hypothesized to be involved in the immunopathogenesis of OLP are:
- Antigen-specific mechanism.
- Non-specific mechanisms.
- Autoimmune response.
- Humoral immunity.
Antigen-specific mechanism:
It has been stated that OLP is a T-cell mediated autoimmune disease in which the auto-cytotoxic CD8+ T-cells trigger apoptosis of the basal cells of the oral epithelium 22. The exact antigen related to lichen planus is still …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
The recognition of self-antigen results in movement of T-lymphocytes into the epithelium. The activated T-cells kill the keratinocytes resulting in ‘liquefaction degeneration’. The keratinocyte apoptosis may result from T-cell secreted TNF-α binding to TNF-α R1 receptor on the keratinocyte surface or binding of T-cell surface CD95L (Fas ligand) to CD95 (Fas) on the keratinocyte surface or granzyme B-mediated cell death.

Non-specific mechanisms in oral lichen planus:
Various non-specific mechanisms have been explained which may result in the development of OLP by facilitating lymphocyte migration into the epithelium and resultant cellular damage. These factors include the epithelial basement membrane, matrix metalloproteinases, chemokines and mast cells. Keratinocytes secrete collagen Type IV and laminin V in the basement membrane zone, which are required for the maintenance of the basement membrane. On the other hand, a normal basement membrane is required for keratinocyte survival. Apoptosis of keratinocytes may result in the disruption of the basement membrane which may in turn, thereby allow the non-specific T-cells to infiltrate the epithelial cell layers 24.
Concentrations of MMP-9 have been found to be increased in culture supernatants taken from patients with OLP as compared to normal controls 25. MMP-9 is a gelatinase that cleaves collagen Type IV. This may disrupt the …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Mast cells are increased in OLP lesion. Not only they are increased in number, but they also degranulate. The degranulation of mast cells leads to the secretion of various pro-inflammatory mediators, including TNF-α, chymase, and tryptase. TNF-α promotes lymphocytes to adhere to the luminal surfaces of blood vessels and their subsequent extravasation. Chymase is an activator of MMP-9 which results in the basement membrane damage. Both TNF-α and chymase stimulate CCL5 secretion by lesional T-lymphocytes, which in turn can trigger further mast cell degranulation. Hence, all these interrelated mechanisms result in lymphocyte infiltration in the epithelium 26.
Autoimmune response:
Many features of OLP support its autoimmune etiology. These include chronicity of the disease, adult onset, female predilection, association with other autoimmune diseases, occasional tissue-type associations, depressed immune suppressor activity in OLP patients, and the presence of auto-cytotoxic T-cell clones in LP lesions 19, 20.
Humoral immunity:
The exact role of humoral immunity in the etiopathogenesis of OLP is not clear, but circulating antibodies have been identified in OLP patients, including autoantibodies against desmogleins 1 and 3 19, 20.
Histology of oral lichen planus:
The histological examination of OLP reveals hyperkeratosis, degeneration of the basal epithelial layer, intense band-like lymphocytic infiltrate of the connective tissue and saw-toothed rete ridges. In atrophic epithelium, rete ridges may be shortened and pointed or totally absent. The degenerating basal keratinocytes result in ‘liquefaction degeneration’ in the basal cell layer. Eosinophilic colloid bodies (Civatte bodies) are formed by degenerating basal keratinocytes and immunocomplexes, and they are often identified in the suprabasal epithelial area 27. The ultrastructure of colloidal bodies suggests that they are apoptotic keratinocytes. Due to the degeneration of keratinocytes and weakening of basement membrane and basal keratinocyte anchoring elements, epithelial-connective tissue interface may show a histological cleft formation (Max Joseph space). Any signs of epithelial dysplasia are absent. B-cells and plasma cells are infrequent in OLP and immunoglobulin and complement deposits are not a consistent feature. Perivascular lymphocytic infiltration is not typically present, as opposed to lupus erythematosus and lichenoid reactions.
Some cases show fibrinogen and fibrin deposition in a linear pattern in the basement membrane zone. In areas of heavy fibrin deposition and colloid body formation, laminin and fibronectin staining may be absent, suggesting basement membrane damage in these areas. The WHO (1978) histological criteria for the diagnosis of OLP includes,
1. Dense, band-like chronic lymphocytic infiltrate beneath and partly inside the basal cell layer.
2. Liquefactive degeneration of the basal cells.
3. Civatte bodies (colloid bodies).
4. Saw-tooth appearance of rete pegs.
5. Acanthosis or epithelial atrophy.
6. Hyperkeratosis, parakeratosis.
Immunofluorescence:
Immunofluorescent findings in OLP are not diagnostic 21. The direct immunofluorescence demonstrates the presence of fibrinogen in the basement membrane zone in 90-100% of the cases 28-31. In some cases, deposition of IgM, and less often IgA, IgG and complement C3, were found exclusively on the colloid bodies. Indirect immunofluorescence is used to detect antibodies that are circulating in the blood. In the diagnosis of OLP, indirect immunofluorescence is not a useful technique either alone or as an adjunct to other diagnostic methods. However, some studies have indicated its use in the diagnosis of lichenoid drug reactions 32, 33.
Differential diagnosis:
OLP should be differentiated from the following conditions,
- Pemphigoid vulgaris.
- Bullous pemphigoid.
- Cicatricial pemphigoid.
- Paraneoplastic pemphigoid.
- Linear IgA bullous dermatosis.
- Epidermolysis bullosa acquisita.
- Dermatitis herpetiforme.
Treatment:
Most of the patients with OLP are asymptomatic. Only the erosive and atrophic OLP lesions are painful and possess a risk of malignant transformation. Many drugs have been used for the treatment of OLP (Figure 26.5). Corticosteroids are the most commonly used drugs for the treatment of OLP. The rationale behind their usage is their ability to modulate inflammation and immune response. They act by reducing the lymphocytic exudate and stabilizing the lysosomal membrane 34. Topical application of mid-potency corticosteroids such as triamcinolone acetonide, high-potent fluorinated corticosteroids such as …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
The non-pharmacological modalities used for the treatment of LP are PUVA therapy (long wave ultraviolet light application), photodynamic therapy (PDT) and laser therapy
Pemphigus
The group of pemphigus diseases includes dermatological disorders characterized by the formation of intra-epithelial blistering. It is an autoimmune disorder that is most prevalent between the 4th and 6th decades of life. Pemphigus can be classified into six types: pemphigus vulgaris, pemphigus vegetans, pemphigus erythematosus, pemphigus foliaceus, paraneoplastic pemphigus, and IgA pemphigus.
Pemphigus vulgaris
It is a painful autoimmune disease in which the patient forms antibodies against desmogleins (Dsg), which are cadherin-type cell to cell adhesion molecules found in the desmosomes 41. The antibodies inhibit the adhesive function of desmogleins (Dsg), resulting in blister formation due to the loss of cell to cell adhesion of keratinocytes (acantholysis). In this condition IgG (IgG1 and IgG4) antibodies and formed against desmoglein 1 and desmoglein 3, resulting in mucocutaneous involvement. If these antibodies are formed against desmoglein 3, there is exclusive mucosal involvement 42. The cutaneous involvement can be severe, generalized skin lesions may lead to septicemia, fluid loss, and electrolyte imbalance. The condition becomes more debilitating if there is involvement of the genitalia and eyes 43. The peak incidence of pemphigus vulgaris occurs between the fourth and sixth decades of life with a male-to-female ratio of I : 2 44. Indirect immunofluorescence or enzyme-linked immunosorbent assay (ELISA) are used to detect serum anti-Dsg1 and anti-Dsg3 IgG autoantibodies for the diagnosis of this condition.
Oral lesions:
The oral lesions appear most commonly on the soft palate, buccal mucosa, and lips. Small vesicles or bullae filled with fluid are present, but there is no inflammation. When the epithelial wall of the vesicle or bulla rapture and becomes detached, a flat painful lesion arises. Lesions are either uncovered or covered with whitish fibrin pellicles. When lateral pressure is applied to the mucosa, a bulla similar to that on the skin is formed, demonstrating a positive Nikolsky’s sign. In some cases, the oral lesions have an atypical appearance and chronic course. In other cases, oral lesions show clinical and morphological features of aphthae which may appear at any site in the oral cavity, especially in areas where the mucosa is strongly traumatized and mechanically strained. The oral lesions of pemphigus vulgaris may also be in the form of flat erosions, which may be easily misdiagnosed as denture sores in the denture wearing patients. In such cases, the diagnosis on the basis of clinical findings is difficult to make. Definitive tests such as indirect immunofluorescence or enzyme-linked immunosorbent assay (ELISA) are done to confirm the diagnosis. Patients usually complain of increased salivation and problems with chewing and swallowing. This condition may result in gradual weight loss and the patient may become cachectic.
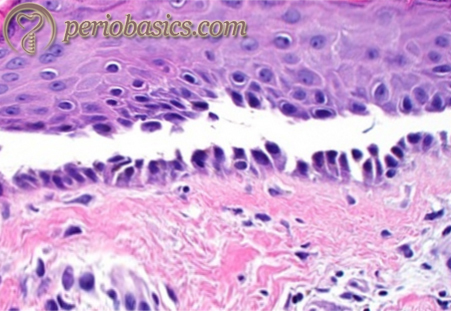
Histopathology:
The histopathological examination shows intraepithelial blister formation associated with extensive acantholysis of keratinocytes 45, 46. The blisters often form, leaving one basal layer at the bottom; such blistering is described as “tombstone-like” (Fig 26.6). There is intercellular edema in the suprabasal portion of the stratum spinosum. The acantholytic cells floating in the stratum spinosum can be seen. Connective tissue stroma demonstrates loose connective tissue network with dense inflammatory cells. Histo-pathologically, pemphigus vulgaris can be differentiated from pemphigus foliaceus. In pemphigus vulgaris, the separation of keratinocytes occurs in the lowest portion of the epidermis, usually seen as the space between the basal cells (those attached to the basement membrane) and the cells above (creating a “tombstone-like” appearance). In contrast, the histologic blister produced by pemphigus foliaceus is most often seen as a split between the most superficial keratinocytes, usually within the granular layer of the epidermis, just beneath the stratum corneum.
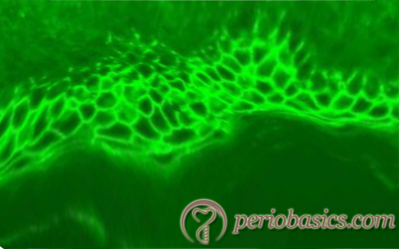
Immunofluorescence:
Direct immunofluorescence of pemphigus vulgaris shows IgG deposition on the cell surface of epidermal keratinocytes. In the direct immunofluorescence, a fluorescent-labeled antibody is used to detect the antigen, which then can be observed by fluorescent microscopy of the tissue section. In the case of pemphigus vulgaris, this antigen-antibody reaction takes place intracellularly in the epithelium, creating a distinct immunofluorescent appearance referred to as “chicken wire effect”. The circulating antibodies against desmogleins can be detected in patient’s serum, using enzyme-linked immunosorbent assay (ELISA).
Differential diagnoses:
The differential diagnosis of pemphigus vulgaris includes the following diseases,
- Bullous Pemphigoid.
- Dermatitis Herpetiformis.
- Erythema Multiforme.
- Familial Benign Pemphigus (Hailey-Hailey Disease).
- Linear IgA Dermatosis.
- Pemphigus Erythematosus.
- Pemphigus Foliaceus.
- Pemphigus Herpetiformis.
- Pemphigus, Drug-Induced.
- Pemphigus, IgA.
- Pemphigus, Paraneoplastic.
- Mucous membrane pemphigoid.
Treatment:
Corticosteroids are the primary therapeutic agents used in the management of pemphigus vulgaris. After the initiation of corticosteroid therapy, clinical improvement may be seen within a few weeks. On an average, cessation of blistering takes 2-3 weeks 47-49 and full healing may take 6-8 weeks 50. The suggested dosage of corticosteroids is: in mild disease, the initial prednisolone doses are 40-60 mg/ day and in more severe cases, 60-100 mg/day. If there is no response within 5-7 days, the dose should be increased in 50-100% increments until there is disease control, i.e. no new lesions and healing of the existing ones. If the dosage of >100 mg per day is required, pulsed intravenous corticosteroid administration could be considered 51. In patients with oral lesions, measures such as soft diets and soft toothbrushes help to minimize local trauma. Prior to eating or toothbrushing, the application of topical analgesics or anesthetics, for example, benzydamine hydrochloride helps in alleviating oral pain. Mouthwashes such as chlorhexidine 0.12 or 0.2% are advised to the patient. Patients using topical corticosteroids are at a risk of developing oral candidiasis which should be treated using antifungal agents.
Adjuvant drugs having steroid-sparing action are also commonly used with the aim of increasing efficacy of the treatment. These drugs include azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate, cyclosporin, tetracyclines/nicotinamide, dapsone/sulphonamides and chlorambucil. Treatment with intravenous immunoglobulin (IVIG) has been reported to cause clinical improvement in patients with pemphigus vulgaris 52-61. The proposed mechanisms of action of these immunoglobulins include blockage of Fc receptors, alteration of cytokine production, cytokine antagonism, inhibition of complement-mediated damage, and neutralization of circulating autoantibodies 62, 63.
Pemphigoid
Pemphigoid is a family of diseases which includes conditions such as bullous pemphigoid which generally affect the skin and have only minor oral involvement and cicatricial pemphigoid which mainly involves the mucous membranes most frequently, the ocular and oral mucosa 64. These diseases are characterized by the formation of the immune-mediated subepithelial split.
Bullous pemphigoid (BP)
In BP, an autoimmune response is generated against two components of the hemidesmosomes of stratified epithelia, referred to as BP 230 kDa (BPAg1) and BP 180 kDa (BPAg2). BPAg1 is a cytoplasmic protein involved in the anchorage of intermediate filaments to the cytoskeleton. BPAg2 is a transmembrane adhesion molecule with several collagenous extracellular domains. Antibodies to BPAg2 appear to be important in subepidermal blister formation. BPAg1 may have a secondary role, and its exact role in the pathogenesis of BP is not fully defined 65. Chromosomal mapping studies localized the BP230 gene to the short arm of chromosome 6, locus 6p11-12 66 and the BP180 gene to the long arm of chromosome 10, locus 10q24.3 67. Autoantibodies against alpha 6 integrin and laminin-5, two other skin basement membrane components, have also been identified in this condition 68.
The exact cause of autoimmune response against BP antigens is not fully understood. It is likely that BP is a result of the …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Oral findings:
Oral lesions are rare, predominantly affecting the buccal mucosa. The bullae usually heal with post-inflammatory pigmentary changes and there is no scarring.
Histopathology:
The histopathological examination demonstrates the subepidermal blister. The inflammatory infiltrate is typically polymorphous, with an eosinophil predominance. Peri-vascular lymphohistiocytic infiltrate is observed. The underlying dermis is inflamed and, usually, shows widespread severe edema. A typical finding in bullous pemphigoid is the retention of the dermal papillary outline (festooning) which project like sentries into the vesicle cavity. Eosinophilic spongiosis is also sometimes evident in the adjacent epidermis.
Immunofluorescence:
BP is diagnosed by direct and indirect immunofluorescence tests. A biopsy is taken which includes a portion of the blister and normal skin. The direct immunofluorescence test is done to identify the deposition of autoantibodies and complement components.
Direct immunofluorescence test:
The most reliable diagnostic criteria of BP are the linear deposition of IgG and C3 at the basement membrane zone (BMZ) and the frequent presence of circulating IgG BMZ antibodies. Direct immunofluorescence tests usually demonstrate IgG (70-90% of patients) and complement C3 deposition (90-100% of patients) in a linear band at BMZ. However, this pattern of immunoreaction is not specific for BP and may be seen in cicatricial pemphigoid and epidermolysis bullosa acquisita. To differentiate BP from other conditions, the patient’s skin biopsy is incubated in 1 mol/L salt solution prior to performing the direct immunofluorescence technique. This process induces cleavage through the lamina lucida. Direct immunofluorescence on salt-split skin reveals IgG on the blister roof (epidermal side of split skin) in patients with BP, while in cicatricial pemphigoid and epidermolysis bullosa acquisita, the IgG localizes to the blister floor (dermal side of split skin).
Indirect immunofluorescence test:
This test is used to identify the presence of IgG circulating autoantibodies in the patient’s serum that target the skin basement membrane component. Approximately 70% of BP patients demonstrate circulating autoantibodies. The titer of circulating antibodies is not correlated with the disease course.
Other tests:
Enzyme-linked immunosorbent assay (ELISA):
This test is used to identify the presence of BP antigen-specific IgG autoantibodies in the patient’s sera. Using various lengths of recombinant proteins of the BPAg1 or BPAg2 antigens, antibodies in serum are identified which is diagnostic of BP.
Direct and indirect immunoelectron microscopy:
These tests are used to identify the binding site of antibodies in the basement membrane zone. The direct immunoelectron microscopy ultrastructurally localizes in vivo- bound IgG autoantibodies whereas indirect immunoelectron microscopy recognizes the binding site of circulating IgG autoantibodies at the basement membrane. In BP, IgG antibodies are detected at the hemidesmosome/upper lamina lucida areas of the basement membrane.
Immunoblotting:
Immunoblotting or the Western immunoblotting test is used to identify the reactivity of IgG derived from the patient’s serum with proteins extracted from the healthy human skin. The sensitivity of this test is variable. In about 75% of cases, these antibodies react with the BP230 antigen, while in about 50% of cases these antibodies react with the BP180 antigen.
Differential diagnosis:
The differential diagnosis of bullous pemphigoid includes the following conditions,
- Cicatricial pemphigoid.
- Dermatitis herpetiformis.
- Drug-induced bullous disorders.
- Epidermolysis bullosa.
- Epidermolysis bullosa acquisita.
- Erythema multiforme.
- Linear IgA dermatosis.
Treatment:
BP is usually a self-limiting disease with a clinical course that may last from months to years. The disease is associated with significant morbidity during the active phase. Older age at onset and frail general condition are poor prognostic factors. The primary aim of the treatment is to control the symptoms and minimize pain.
Corticosteroid therapy is the first line of drugs in the treatment of BP. Localized BP often can be treated successfully with topical steroids alone. More extensive disease, which is often more difficult to control, is usually treated with systemic anti-inflammatory and immunosuppressive agents, oral corticosteroids being the mainstay of treatment. Oral prednisone/ prednisolone doses range from 0.3 to 1.25 mg/kg body weight/day, which usually controls the disease within 1-2 weeks; the dose is then progressively tapered.
Adjuvant drugs used in the treatment of BP are azathioprine, antibiotics with anti-inflammatory effects (doxycycline, minocycline, lymecycline), methotrexate, mycophenolate mofetil, dapsone, and sulfonamides. Intravenous immunoglobulin (IVIG) is another immuno-modulatory treatment which has been successfully used in BP patients 52, 55, 59, 70.
Cicatricial pemphigoid
Cicatricial pemphigoid (mucous membrane pemphigoid or ocular or oral-gingival pemphigoid) is a rare, blistering disease of the skin, characterized by severe, erosive lesions of the skin and mucous membranes. The term cicatricial is derived from the word ‘cicatrix’ meaning a scar, as it heals with scarring 71. This condition is also recognized by the term “immune-mediated subepithelial blistering diseases” because it involves autoimmune subepidermal/subepithelial split. In this condition, autoantibodies are directed against one or more components of the basement membrane. These include bullous pemphigoid antigen 1 (BPAg1, 230 kd) 72, bullous pemphigoid antigen 2 (BPAg2, 180 kd) 73, laminin 5 74, laminin 6 75, α6-integrin subunit 76, β4-integrin subunit 77, and collagen Type VII 78. This disease usually occurs in …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Oral lesions:
Oral lesions mostly manifest as scattered painful erosions or desquamative gingivitis with smooth erosions along the attached gingiva or rarely mucosal detachment. Scars may be present, but oral blisters are rare 79. The bullae usually rupture after 2-3 days of formation, leaving behind irregularly shaped areas of ulceration. Healing of the lesions may take up to 3 weeks or longer. Due to buccal mucosal scarring, the patient may complain of difficulty in chewing and swallowing.
Ocular lesions:
The ocular lesions in cicatricial pemphigoid include the recurrent conjunctival inflammatory process that usually results in subepithelial fibrosis, leading to fornix shortening, symblepharon, and ankyloblepharon formation, and then subsequently trichiasis and entropion. The scarring of the conjunctiva also results in loss of goblet cells with subsequent dry eye and disruption of the corneal epithelium 80. Ocular ulcerations that heal with scaring may lead to blindness.
Histopathology:
The histopathological examination cannot completely diagnose mucous membrane pemphigoid. However, it clearly reveals a subepithelial blister with a variable inflammatory cell infiltrate. The separation of the epithelium and the connective tissue occurs at the basement membrane zone.
Immunofluorescence:
Direct immunofluorescence shows a linear deposit of IgG, IgA, C3 or a combination of these in the basement membrane zone 81. However, this finding is common to various forms of pemphigoid and other related diseases. For an accurate diagnosis, more sophisticated investigation using direct immunofluorescence is required. This includes use of salt-split skin or computer-aided fluorescence overlay antigen mapping with laser scanning confocal microscopy 82-84. Indirect immunofluorescence can be used to identify circulating autoantibodies. However, all the patients with mucous membrane pemphigoid do not have identifiable autoantibodies. Only 60-65% of patients are positive for circulating auto-antibodies 85.

Differential diagnoses:
Differential diagnoses of mucous membrane pemphigoid include the following diseases,
- Pemphigus vulgaris.
- Paraneoplastic pemphigus.
- Bullous pemphigoid.
- Bullous lichen planus.
- Dermatitis herpetiformis.
- Linear IgA.
- Erythema multiforme.
- Epidermolysis bullosa acquisita.
- Stevens‐Johnson syndrome.
Treatment:
The treatment of mucous membrane pemphigoid depends on the severity of the disease. In the case of mild disease, the drug of choice is dapsone, which may be initiated at a dose of 25 to 50 mg per day, increasing monthly by 25 to 50 mg until clinical remission is achieved or until the maximum tolerated dose is reached (usually 200 mg per day) 86. The main adverse effects of the treatment with dapsone include hemolysis, methemoglobinemia, agranulocytosis, and peripheral neuropathy. It is therefore recommended that a complete blood count with differential reticulocyte count be done every 2 weeks for the first 3 months of therapy and then every subsequent 3 months 87. This drug is contraindicated in patients with G6PD- deficiency. In certain patients, only oral lesions may be present. Successful treatment of the oral lesions in mucous membrane pemphigoid has been reported with the use of topical steroids, tacrolimus, and cyclosporine 88-93.
In severe disease, various systemic drugs have been shown to be useful in the treatment of mucous membrane pemphigoid. Systemic corticosteroids are the agents of choice for the initial treatment, combined with steroid-sparing agents for long-term maintenance. Steroid-sparing drugs include cyclophosphamide, azathioprine, methotrexate, and mycophenolate mofetil. For patients who do not respond appropriately to these drugs, the treatment of choice is high-dose intravenous immunoglobulin (IVIG) administration. As already stated, the proposed mechanisms of IVIG action are blockage of Fc receptors, alteration of cytokine production, cytokine antagonism, inhibition of complement-mediated damage, and neutralization of circulating autoantibodies 62, 63.
Linear IgA disease
Linear IgA disease is a rare autoimmune vesiculobullous disorder characterized by linear IgA deposits at the basement membrane zone (BMZ) on immunofluorescence. The clinical features of this disease are difficult to distinguish from dermatitis herpetiformis. The distinction is made on the basis of immunopathologic findings and the absence of an associated gluten-sensitive enteropathy 94. This disease is caused due to IgA autoantibodies typically directed against the components of hemidesmosomes and basement membrane (BP180, BP230, and LAD285).
Linear IgA bullous disease affects adults of all ages, but is most commonly seen in the patients older than 30 years 95. Chronic bullous disease of childhood occurs in young children, usually presenting in those younger than five years 96. The lesions of linear IgA dermatosis consist of pruritic, annular papules, vesicles, and bullae that are found in groups. Vesicles or bullae are often target-shaped, surrounding an erythematous area of skin which is flat or raised. Lesions have a predilection for the extensor surfaces, with symmetric distribution.
Linear IgA bullous disease usually arises spontaneously, but sometimes, infections and occasionally medicines may trigger this disease. Sometimes other underlying causes may also be associated with this disease. Linear IgA bullous dermatosis has been reported in patients with inflammatory bowel disease. It is thought that autoantigens are exposed to the immune system in the diseased colon, and the same autoantigens are then targeted in the skin. Surgery to remove bowel can lead to clearance of this blistering skin disease 97.
Oral lesions:
Oral ulcers are noted in 50 % of the cases 96. The hard and soft palate are more commonly affected. Oral lesions are painful and erosions and ulcerations can be seen. Rarely, oral lesions may be the only clinical manifestation of linear IgA disease and may precede the cutaneous manifestations.
Histopathology:
The histopathological findings are very similar to those found in the case of erosive OLP.
Immunofluorescence:
Usually, the definitive diagnosis is made by direct immunofluorescence analysis, revealing a homogenous linear pattern of IgA deposition in the basement membrane zone. Occasionally, direct immunofluorescence also will be weakly positive for IgG and/or IgM.
Differential diagnosis:
Other conditions which have an almost identical appearance include:
- Bullous pemphigoid.
- Dermatitis herpetiformis.
- Lupus erythematosus.
- Epidermolysis bullosa acquisita.
Treatment:
Sterile dressings should be used to cover the ruptured lesions and erosions to prevent any infection. Mild disease can be treated with topical corticosteroids. Dapsone or sulfapyridine are the treatments of choice for non drug-related dermatitis. Other medications such as steroids, cyclosporine, mycophenolate mofetil and colchicines may be used.
Lupus erythematosus (LE)
LE is a heterogeneous, multisystem, autoimmune disease characterized by the production of autoantibodies against several cell constituents, occurring predominantly in young women. Systemic lupus erythematosus (SLE) and cutaneous lupus erythematosus (CLE) are the main clinical variants of this disease. The primary pathological findings in patients with SLE are those of inflammation, vasculitis, immune complex deposition, and vasculopathy. SLE most commonly affects the heart, joints, skin, lungs, blood vessels, liver, kidneys, and nervous system. The skin is involved in up to 85% of SLE cases and may be the only organ involved in CLE.
The clinical manifestations of SLE are fundamentally the same in children and adults. However, in childhood-onset SLE, there are several clinical symptoms more commonly found than in adults, including malar rash, ulcers/ mucocutaneous involvement, renal involvement, proteinuria, urinary cellular casts, seizures, thrombocytopenia, hemolytic anemia, fever, and lymphadenopathy 98. Mortality rates for SLE are particularly high in children. The diagnosis of the cutaneous manifestations of LE is based on the clinical appearence, histopathology, and immunohistology of the skin lesions. Based on clinical morphology and specific histological findings, the CLE can be classified as 99,
1. Acute cutaneous lupus erythematosus (ACLE).
2. Subacute cutaneous lupus erythematosus (SCLE).
3. Chronic cutaneous lupus erythematosus (CCLE),
> Discoid lupus erythematosus (DLE),
a. Hypertrophic/verrucous variant.
b. Teleangiectoid variant.
> Lupus erythematosus profundus (LEP).
> Chilblain lupus erythematosus (CHLE).
The cutaneous manifestations of LE include erythematous patches on the face, which coalesce to form a roughly symmetrical pattern over the cheeks across the bridge of the nose in a butterfly-like distribution. In some cases, areas of hyperpigmentation may be seen. The skin over the neck, arms, shoulders, and fingers are also affected. Itching or burning sensation may also be associated with the affected areas.
Etiopathogenesis:
The exact etiology of LE remains to be clarified. It is believed that the interactions between genetic, infectious and hormonal factors may alter the immunoregulatory mechanism, resulting in the generation of an autoimmune response against a variety of self-antigens. All these factors cause increased …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Oral findings:
Oral lesions of SLE develop in 20-50% of the patients, presenting as painless, shallow oral ulcers, most often occurring on the hard and soft palate. The classic presentation is the oral discoid lesion, characterized by a well-demarcated zone of erythema, atrophy, or ulceration surrounded by white, radiating striae. These have clinical similarity with erosive oral lichen planus lesions 103. The lesions appear as maculae (red patches) that will later transform into irregular erosions and ulcers which often heal with scarring. Purpuric lesion such as ecchymosis and petechiae may occur. Discoid lesions like those typically found elsewhere on the sun-exposed skin may be found on the lip vermilion. In some cases, cheilitis may also be present 104.
Diagnosis:
The diagnosis of LE is made based on the clinical features and serological tests. As stated earlier, SLE is characterized by the formation of autoantibodies against self-antigens. Some of these can be used as diagnostic markers, while others can help in quantifying the disease activity. A brief description of these tests is as follows,
Antinuclear antibody (ANA) test:
ANA is a standard screening test for SLE because 95% of cases show a high titer (1:80 or more) of this autoantibody. ANAs are actually a family of autoantibodies, which may be directed against any one of the nuclear antigens, including double-stranded DNA, extractable nuclear antigens (ENA), histones and nuclear RNA. However, it must be remembered that this test is positive in some other conditions also such as systemic sclerosis, Sjogren’s syndrome, overlap syndrome, antiphospholipid syndrome, polymyositis and rheumatoid arthritis. Many laboratories use ELISA technique for the detection of ANA due to convenience and economy. However, the gold standard for testing and reporting ANA is the indirect immunofluorescence method.
Anti-double stranded DNA antibody (anti-dsDNA) test:
This test specifically targets double-stranded DNA antibodies. However, the technique must ensure the absence of any contamination with single-stranded DNA in the antigen used. A positive anti-dsDNA is highly suggestive of SLE. Also, anti-dsDNA titers most often correlate with the disease activity.
Antibodies to extractable nuclear antigens (anti-ENA):
The extractable nuclear antigens include anti-Sm, anti-UIRNP, anti-Ro and anti-La antibodies. Anti-ENA antibodies are found only in about 50% of sera which are positive for ANA.
Anticardiolipin (aCL) antibodies and lupus anti-coagulant:
The anticardiolipin antibodies mostly belong to IgG class, however, they may be present along with IgM antibodies. These can be detected in patient’s serum using the ELISA test. Similarly, lupus anticoagulant is also associated with SLE.
Complement levels (C3 and C4):
These two complement components are useful in the diagnosis of SLE because their serum levels are associated with lupus activity. Their levels in serum are reduced due to their consumption and are negatively correlated with lupus activity. The American College of Rheumatology has given the criteria for the classification of patients having SLE 105. If a patient has, at any time in his or her medical history, 4 of the 11 criteria documented, the diagnosis of SLE can be made with about 95% specificity and 85% sensitivity.
| Clinical feature | Definition |
|---|---|
| Malar rash | Fixed erythema, flat or raised, over the malar eminences, sparing nasolabial folds. |
| Discoid rash | Erythematous, raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions. |
| Photosensitivity | Skin rash as a result of unusual reaction to sunlight by history or on physical exam. |
| Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by the physician. |
| Non-erosive arthritis | Involving two or more peripheral joints, characterized by tenderness, swelling or effusion. |
| Pleuritis/pericarditis | a. Pleuritis- convincing h/o pleuritic pain or rub or pleural effusion on physical examination. OR b. Pericarditis- documented by ECG, rub or e/o effusion. |
| Renal disorder | a. Persistent proteinuria > 0.5 gm/day or > +++, OR b. Cellular casts- may be red cell, Hb, granular, tubular or mixed. |
| Neurological disorder | a. Seizures- in the absence of offending drugs, or known metabolic derangement, e.g. uremia, ketoacidosis or electrolyte imbalance, OR b. Psychosis- in the absence of offending drugs, or known metabolic derangement, e.g. uremia, ketoacidosis or electrolyte imbalance. |
| Hematological disorders | a. Hemolytic anemia with reticulocytosis, OR b. Leukopenia < 4000/cu mm on 2 or more occasions, OR c. Lymphocytopenia < 1500 on 2 or more occasions, OR d. Thrombocytopenia < 100,000/cu mm in the absence of offending drugs. |
| Immunological disorder | a. Anti-DNA: antibody to native DNA in abnormal titer, OR b. Anti-Sm (Smith) antibodies: presence of antibody to Sm nuclear antigen, OR c. Positive finding of antiphospholipid antibodies based on 1) a serum level of IgG or IgM anticardiolipin antibodies or 2) a positive test result for lupus anticoagulant, using a standard method, or 3) a false-positive test for syphilis for at least 6 months and confirmed by TPI or FTA-abs test. |
| Positive ANA | An abnormal titer of ANA by immunofluorescence or an equivalent assay at any point in time in the absence of drug. |
Histopathology of oral lesions:
The histopathological investigation demonstrates para-keratosis, hydropic degeneration of stratum germinativum, collagen degeneration, and a lymphocytic infiltration in a perivascular orientation. The lamina propria exhibits a chronic inflammatory cell infiltrate similar to that observed in lichen planus.
Immunofluorescence:
Direct immunofluorescence findings show immunoglobulin (mostly, IgM) deposition at the basement membrane zone or at colloid bodies. C3 deposits are seen at the dermal-epidermal junction. As discussed earlier, the indirect immunofluorescence reveals the presence of ANAs in more than 95% of the patients.
Differential diagnosis:
The following conditions need to be considered in the differential diagnosis of SLE
- Undifferentiated connective tissue disease.
- Primary Sjogren’s syndrome.
- Primary antiphospholipid syndrome.
- Fibromyalgia with positive ANA.
- Idiopathic thrombocytopenic purpura.
- Drug-induced lupus.
- Early rheumatoid arthritis.
- Systemic vasculitis.
Treatment:
Management of SLE often depends on the individual patient’s disease severity and disease manifestations. Hydroxy-chloroquine has a central role in the long-term treatment of all SLE patients. According to the severity of the disease and requirement, following group of drugs are used,
Biologic DMARDs (disease-modifying antirheumatic drugs): Belimumab, rituximab, iv immunoglobulins.
Nonbiologic DMARDS: Cyclophosphamide, methotrexate, azathioprine, mycophenolate, cyclosporine.
Nonsteroidal anti-inflammatory drugs (NSAIDS): e.g. Ibuprofen, naproxen, diclofenac.
Corticosteroids: e.g. Methylprednisolone, prednisone.
Antimalarials: e.g. Hydroxychloroquine.
Erythema multiforme (EM)
EM is an acute mucocutaneous disorder, characterized by varying degrees of blistering and ulceration. The disease is characterized by skin eruptions, with or without oral or other mucous membrane lesions 106-108. “Target” or “iris” lesions with central clearing, are the hallmark of EM. EM can be classified as erythema multiforme minor, erythema multiforme major, Stevens-Johnson syndrome and toxic epidermal necrolysis, where, erythema multiforme minor is the mildest type of lesion and toxic epidermal necrolysis is the most severe form 107, 108. EM minor shows ulcerations involving a single mucosal site with typical skin target lesions. EM major shows ulcerations involving more than one mucous membrane with skin target lesions. Classic EM is a self-limiting skin disease. However, some patients have frequent episodes over the years (recurrent EM) and, rarely; others may have the continuous uninterrupted disease (persistent EM).
Etiopathogenesis:
Many etiological factors have been associated with EM. There is a large body of evidence suggesting infections and drug usage as the main etiological factors in the initiation of this disease. Other factors proposed as etiological factors for the disease include radiation, menstruation, autoimmune diseases, and malignancies 109. Herpes simplex virus (HSV) type 1 is most commonly associated with EM, however, HSV type 2 and Mycoplasma pneumoniae are also important causes of the condition 109, 110. Along with this, certain immunizations and other immune-related conditions, food additives and chemicals are also reported to be triggering factors of EM.
The pathogenesis of herpes-associated erythema multi-forme (HAEM) is consistent with a delayed-type hyper-sensitivity reaction. The cell-mediated response is directed against viral antigen positive cells that contain the HSV DNA polymerase gene (pol). This herpes simplex virus pol DNA is located in the …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Oral lesions:
Around 25-50% of all EM minor patients have oral manifestations 115, 116. The oral lesions are usually erythematous macules on the lips and buccal mucosa, followed by epithelial necrosis, bullae, and ulcerations with an irregular outline and a strong inflammatory halo. Hemorrhagic crusting of the lips and ulceration, mainly of the non-keratinized mucosa, is usually evident. The intraoral mucosal lesions are mainly present on the anterior part of the oral cavity, the tongue and the buccal mucous membrane 117. Due to erosions of the oral mucosa patient may have difficulty while eating, drinking, or opening the mouth.
Histopathology:
The histopathological examination aids in the diagnosis of EM. Histological findings demonstrate liquefactive degeneration of the basal epidermal cells, necrotic keratinocytes, and lymphocyte exocytosis. A dense lymphohistiocytic infiltrate can be seen at the dermo-epidermal junction as well as around the blood vessels.
The final diagnosis should be made on the basis of clinical findings, combined with histological findings. Specific questions should be asked about HSV, M. pneumoniae, or other infections, and the use of any new medications.
Differential diagnosis:
There are many clinical conditions which should be differentiated while diagnosing EM. These include,
- Herpes simplex virus (HSV) infection
- Mycoplasma pneumoniae infection.
- Acute generalized exanthematous pustulosis.
- Collagen vascular diseases.
- Disseminated lesions of contact dermatitis, exfoliative dermatitis.
- Erythroderma.
- Immunoglobulin A (IgA) linear dermatosis.
- Lichen planus.
- Lupus erythematosus.
- Lyme disease.
- Major oral aphthae, recurrent aphthous ulcers.
Treatment:
As already stated, EM is generally regarded as a self-limiting skin disease. The lesions will appear over 3-5 days and resolve over 1-2 weeks. Treatment is based on finding out the cause of the reaction. HSV infection is most commonly responsible for the precipitation of this condition (HAEM). EM precipitates eight days following HSV infection. Any treatment at this point does not alter the clinical course of the disease. However, topical corticosteroids and oral antihistamines can be given to patients with itching and burning of cutaneous lesions. Oral lesions, if present, are painful and cause discomfort to the patient while eating or speaking. High potency topical corticosteroid gel, oral antiseptic washes, and oral anesthetic solutions can be given to these patients. Systemic cortico-steroids can be given, if the oral lesions are very painful. The antiviral therapy for HAEM includes,
Acyclovir- 400 mg BID.
Valacyclovir- 500 mg BID.
Famciclovir- 250 mg BID.
If the antiviral treatment is not able to resolve the condition, the second line of treatment includes azathioprine, dapsone, cyclosporine, or mycophenolate mofetil. Although, controlled clinical trials are required to validate most of these treatments. Tetracycline 250 mg four times a day for at least 1 week may be indicated in EM related to Mycoplasma pneumoniae. If the patient gives a history of intake of any drug which coincides which precipitation of EM, the drug should be discontinued and the physician should be consulted regarding the drug and its substitution, if necessary.
The recurrent EM is difficult to treat. In patients with HSV infection associated recurrent EM and idiopathic recurrent EM, the first-line of treatment is antiviral prophylaxis 110, 118, 119. When the condition does not resolve with antiviral treatment, the second line of treatment includes drugs such as azathioprine, dapsone, mycophenolate mofetil, immunoglobulin, hydroxychloroquine, thalidomide, and cyclosporine.
Dermatitis herpetiformis
Dermatitis herpetiformis or Duhring’s disease is an inflammatory cutaneous disease with a chronic-relapsing course, pruritic polymorphic lesions and typical histopathological and immunopathological findings. It is important to note that despite its name, this disease is neither related to nor caused by herpes virus: the name means that it is a skin inflammation having an appearance similar to herpes. This condition is closely associated with gluten-sensitive enteropathy, but the exact mechanism is unknown 120, 121. It has been proposed that both of these conditions are multifactorial disorders in which genetic and environmental triggering factors play a crucial role.
Males are affected slightly more than females in this disease 122. It occurs commonly between 20-55 years of age; however, this disease has also been reported in children less than 5 years of age 123. The disease follows a chronic relapsing course. This disease is characterized by small, clustered papules and vesicles that erupt symmetrically on the elbows, knees, buttocks, back or scalp. The lesions may be associated with a burning sensation. Older lesions may leave pale or dark marks (hypopigmentation and hyperpigmentation). Flat, red patches, thickened plaques and raised wheals may arise, resembling dermatitis, scabies, and other skin conditions. The small bowel involvement is often asymptomatic in adults; however, it can be associated with abdominal pain, diarrhea, iron deficiency and reduced growth rates in children 46, 124.
Oral lesions:
The oral lesions may be erythematous, pseudo-vesicular, purpuric, or erosive in nature. The small vesicles rupture soon after formation, leaving fibrin-covered, superficial erosions resembling aphthae. Hyperkeratotic areas and lesions resembling oral lichen planus might also occur. Buccal mucosa adjacent to the occlusal plane or area beneath existing dentures is commonly affected.
Etiopathogenesis:
As already stated, a multifactorial etiology has been suggested to explain the etiopathogenesis of this disease. The major environmental factor involved in triggering the disease is exposure to gluten. Gluten is composed of two peptides, gliadin, and glutenin, with the disease pathogenesis being linked to gliadin. Strong association with HLA DQ2 (in 80-90% of patients) and HLA DQ8 (in 10-20% of patients) have been demonstrated in both dermatitis herpetiformis and celiac disease 121, 125, 126. The immunological etiology has been suggested due to …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Histopathological findings:
The histopathological picture of early lesion of dermatitis herpetiformis demonstrates dermal papillary collections of neutrophils, neutrophilic fragments, fibrin, edema, variable numbers of eosinophils, and cleft formation between the dermal papillary tip and the epidermis. The older lesions of dermatitis herpetiformis demonstrate subepidermal bullae that usually cannot be distinguished from those of other subepidermal bullous diseases.
Immunofluorescence:
Direct immunofluorescence of clinically normal skin adjacent to the lesion shows granular IgA deposits in the upper dermis. The IgA deposits in the dermal papillae may appear with different patterns: granular on the papillary dermis, granular on the basement membrane and fibrillar 131. Around 50% of patients show deposition of complement fraction 3 (C3) in dermal papillae 132.
Differential diagnosis:
Other diseases which mimic dermatitis herpetiformis are,
- Atopic dermatitis.
- Papular urticaria.
- Bullous pemphigoid.
- Erythema multiforme.
- Linear IgA dermatosis.
- Scabies.
- Transient acantholytic dermatosis.
Treatment:
Patients with dermatitis herpetiformis should be given a gluten-free diet. It has been seen that in a patient with dermatitis herpetiformis and celiac disease, the gastro-intestinal benefits are observed much earlier than cutaneous ones. Dapsone is the drug of choice in the treatment of dermatitis herpetiformis, with sulfapyridine as the alternative therapy. The exact mechanism of dapsone action is not well understood, but it seems to block chemotaxis and activate neutrophils, besides reducing the release of leukotrienes and prostaglandins. The adult dose of dapsone is 100 mg per day with reduced dose in children. The main side effects of dapsone are acute hemolytic anemia, and methemoglobinemia, which are dose-dependent. Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an absolute contraindication for the use of dapsone as it may cause severe hemolysis.
Epidermolysis bullosa acquisita (EBA)
EBA is an acquired inflammatory and/or dermolytic sub-epidermal blistering disease characterized by IgG auto-antibodies to collagen Type VII (COL7). Various factors have been shown to play an important role in the pathogenesis of this disease. Studies have shown a critical role of the genetic background, T-cells, and cytokines for mediating the loss of tolerance towards COL7 133-136. Altered host immunoregulation resulting in the release of reactive oxygen species, and matrix metalloproteinases are crucial for autoantibody-induced tissue injury in EBA. EBA patients can be classified into two major clinical subtypes: non-inflammatory (classical or mechanobullous) which is characterized by skin fragility, tense blisters, scarring, and milia formation, preferably localized to trauma-prone sites and the extensor skin surface and inflammatory EBA with clinical features ranging from inflammatory subepidermal blisters that are indistinguishable from those in patients with bullous pemphigoid to dermolytic lesions that resemble those seen in patients with dystrophic epidermolysis bullosa 137, 138.
Oral lesions:
The oral lesions are most commonly observed in patients with non-inflammatory EBA. Oral blisters and erosive lesions are observed on the buccal mucosa, tongue, gingiva, palate, and lips. It has been suggested that either perioral or intraoral blistering can lead to microstomia, ankyloglossia, scar formation and obliteration of the oral vestibular area. During blistering and subsequent cicatrization, epithelial cells become entrapped and give rise to milium cysts, particularly in the hard palatal mucosa.
Histopathology:
The histopathological picture shows subepidermal cleavage with marked neutrophilic inflammatory infiltrate and rare eosinophils. A mixed inflammatory cell dermal infiltrate is observed.
Immunofluorescence:
Immunofluorescence is the gold standard for the diagnosis of EBA. The direct immunofluorescence test shows IgG in a linear array along the dermo-epidermal junction. To a lesser extent C3 can also be found deposited linearly at the basement membrane zone. Other immunoreactants such as IgM or IgA may also be seen. Indirect immunofluorescence demonstrates the presence of IgG circulating autoantibodies in the patient’s serum which attack collagen Type VII in the skin basement membrane. This investigation on salt-split normal human skin substrate using serum from the affected patient shows IgG autoantibodies on dermal part (lower part) of the basement membrane. This investigation differentiates EBA from BP because, IgG autoantibodies from patients with BP bind to the epidermal roof (upper part) of salt-split skin. Other conditions where indirect immunofluorescence is detected on the dermal floor are bullous SLE, antiepiligrin cicatricial pemphigoid (with autoantibodies to laminin-5 and laminin-6), and anti-p105 pemphigoid (with autoantibodies to a 105-kd lower lamina lucida protein) 139.
Treatment:
Most EBA patients are treated with systemic corticosteroids with an initial dose ranging from 0.5 to 1.5 mg/kg per day 140. The corticosteroid therapy is combined with other immuno-suppressive/modulatory agents to lower the corticosteroid dose. These agents include methotrexate, azathioprine, and colchicines. High-dose intravenous immunoglobulin (IVIG) has been established as an effective therapy for EBA 141.
Contact stomatitis
Contact stomatitis describes an inflammatory reaction of the oral mucosa caused due to the contact with irritants or allergens. A wide variety of substances are known to elicit adverse oral mucosal reactions. These include aromatic compounds found in chewing gums and toothpastes, most common being carvone, spearmint essential oil, menthol essential oil, cinnamaldehyde, and cinnamon essential oil 142. Allergic contact stomatitis is a hypersensitivity reaction (type IV). The affected individual is sensitized by …… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Histopathological analysis:
The diagnosis can be confirmed with histopathological analysis. The histopathological findings include hyper-orthokeratosis, acanthosis or atrophy and liquefaction degeneration of the basal layer of the affected epithelium. Neutrophil infiltration of the connective tissue is sometimes observed. A perivascular lymphocytic infiltrate may also be present. Eosinophilic infiltration of superficial connective tissue can be seen.
Treatment:
The primary treatment of this disease includes identifying the allergic agent. A carefully recorded case history of the patient plays a very important role in the identification of the offending agent. Analgesics and local anesthetic gels may be used to reduce pain associated with oral ulcerations. A non-spicy bland diet is advised to the patient during the healing period.
Drug-induced oral reactions
Many drugs taken systemically may give rise to numerous adverse oral manifestations, particularly dry mouth, taste disturbances, oral mucosal ulceration, and/or swelling. Oral mucosal ulceration may also be caused as a result of the direct application of over the counter drugs like aspirin, hydrogen peroxide, potassium tablets, and phenol containing compounds. The area in the oral cavity where the drug is kept usually appears whitish and corrugated, with erosion and ulceration 143. The lesion is also referred to as stomatitis venenata. Non-specific oral ulcers may result due to the intake of a variety of drugs. These include barbiturates, β-blockers, dapsone, NSAIDs, phenazone derivatives, thiazide derivatives, phenolphthalein, sulfonamides, tetracyclines, sirolimus etc. 144
Many antineoplastic drugs such as methotrexate, 5-fluoroucil, doxorubicin, and melphalan have oral ulceration as one of their side effects. Oral mucositis is one of the important findings seen in patient undergoing chemotherapy. In most of the cases, there is sloughing and ulceration which can be observed within days of commencement of therapy. The ulceration may be a portal of entry for infection and hence a potential cause of septicemia.
Drug-induced vesiculobullous lesions have also been reported with the use of systemic drugs. Most of them have similar clinical, histopathological and immunofluorescence pattern as seen in pemphigus vulgaris. Thiols are most commonly associated with drug-induced vesiculobullous lesions. They may cause decreased levels of plasminogen activator inhibitor, thereby resulting in increased plasminogen activation and thus epithelial damage. It may result in the exposure of epithelial antigens (e.g., desmoglein 1 and 3), resulting in immunological damage to the skin 145-147.
Linear IgA bullous disease may also result due to drug intake. In this condition, IgA antibodies are directed against several self-antigens, including bullous pemphigoid antigen-I. Many drugs have been reported to induce this condition, including Vancomycin 148. Erythema multiforme and its severe form Steven Johnson syndrome have also been reported to result from drug intake. The cutaneous reaction typically results 1-3 weeks after the ingestion of the drug. Toxic epidermal necrosis has been associated with the intake of antimicrobials, analgesics and allopurinol 149.
Management of drug-induced oral reactions:
The definitive treatment for the drug-induced oral reaction is the discontinuation of the offending drug. However, when the patient has significant discomfort, supportive therapy is given to reduce the intensity of signs and symptoms. Analgesics may be given to control pain. Also, to reduce pain in the ulcerated areas, local application of anesthetic agents such as lignocaine or benzocaine is done. Broad-spectrum antibiotics may be given to minimize the chances of infections. Antifungal ointments may be applied if any fungal infection is suspected. The patient is advised to take soft and bland diet till the complete healing of the ulcers.
Endocrine disturbances causing desquamative gingivitis
Hormonal disturbances may also result in desquamative gingivitis. There is wide evidence that the female sex hormone; estrogen has a protective action on the integrity of the oral mucosa. The significance of female sex hormones in the maintenance of gingival health was recognized as early as 1943, by Richman and Abarbanel 150, 151. Estrogen is synthesized in the ovaries and is a derivative of cholesterol. The three naturally occurring estrogen hormones in females are estrone (E1), estradiol (E2) and estriol (E3). During menopause, the predominant circulating…… Contents available in the book……… Contents available in the book………. Contents available in the book……… Contents available in the book……. Contents available in the book……… Contents available in the book……..
Aging
Aging results in thinning of the oral epithelium and diminished keratinization. Also, flattening of rete pegs and an increase in the height of the epithelial ridges has been shown to be associated with aging. The epithelium-connective tissue interface was investigated in a study using morphological 3-dimensional analysis which showed that connective tissue ridges were observed to be more prevalent in young individuals whereas, connective tissue papillae were predominant in old individuals 154. Although, aging is not a causative factor for desquamative gingivitis but the changes in the epithelium and epithelium-connective tissue interface may contribute to its precipitation.
Chronic infections
Rarely, chronic bacterial, fungal or viral infections may present clinically as desquamative gingivitis. However, there is scanty literature regarding desquamative gingivitis resulting from chronic infections.
Other causes
Rarely, some other systemic disorders may also precipitate gingival ulcerations. These include granulomatous conditions such as orofacial granulomatosis, Crohn’s disease, and sarcoidosis. Gingival ulcerations are also seen in plasma cell gingivitis and chronic ulcerative stomatitis.
Conclusion
It is quite clear from the above discussion that it is very difficult to differentiate between different forms of desquamative gingivitis on the basis of clinical findings. The histopathological and immunofluorescence examinations are often necessary to make an accurate diagnosis. If all these examinations do not lead to a definitive diagnosis, hypersensitivity reactions to oral hygiene products should be suspected. The mainstream therapy of these gingival conditions is still local and systemic corticosteroid therapy. However, a lot of research is going on to find out alternative and curative therapies for different forms of desquamative gingivitis, which may change our approach to treat these diseases.
References
References available in the hard-copy of the website.